- HOME
- CHI SIAMO
- SINDROME DI RETT
- RICERCA
- NEWS
- Margherita, la porta della speranza
- Pasqua 2024 di Pro RETT Ricerca
- Francesca Latella - 21 febbraio 2024
- 16 dicembre a Milano
- Il Natale 2023 di Pro RETT Ricerca
- Azzurra per il 5X1000 2023
- Pro RETT News #07 - marzo 2023
- Pasqua 2023 di Pro RETT Ricerca
- Diventa socio di Pro RETT Ricerca
- Il Natale 2022 di Pro RETT
- Due nuovi progetti nel 2022
- Pasqua 2022
- L'Erbolario sostiene la ricerca scientifica
- Il mercatino di Natale 2021
- Dove c'è un bisogno, c'è un Lion
- Secondo incontro online...
- Incontro online...
- Uniti per accelerare...
- Il Natale 2020 di Pro RETT
- Campagna fotografica 2020
- Pasqua 2020 con Pro RETT
- Le Voci di Rett
- 6th European Conference
- CONTATTACI
Invio in corso, attendere...

Attendi, connessione a PayPal in corso
Digita i caratteri visibili nell'immagine sopra,
nello stesso ordine e senza distinzione tra maiuscole e minuscole.
Digita un indirizzo email valido e in uso,
il nostro staff ti risponderà a questo indirizzo email.
Tutti i prezzi indicati includono IVA
Dopo aver aggiunto un prodotto sarà possibile modificare la quantità nel carrello prima di effettuare l'ordine
Margherita, la porta della speranza
Il libro di Giuditta Mascheri che racconta la sua vita con Margherita
20 marzo 2024
Pro RETT Ricerca è lieta di sostenere e promuovere il libro scritto da Giuditta Mascheri per raccontare la sua esperienza di vita con la figlia Margherita, scomparsa il 21 luglio del 2021 a soli 8 anni.

Margherita, la porta della speranza è – con le parole della stessa Giuditta - "un libro nato come una sorta di diario liberatorio, scritto per lo più nel mese di agosto, quando Margherita ci aveva lasciato da poco ed eravamo in vacanza al mare. Giravo con un diario e una penna. Passavo le ore in spiaggia sotto l'ombrellone o al bar davanti a un caffè a scrivere: ero un po' come un fiume in piena. Avevo bisogno di dare pace alla mia mente che continuava a pensare alla vita passata e temeva di iniziare a dimenticare”.

Presentato ufficialmente lo scorso 7 febbraio 2024 presso l'Istituto Maria Ausiliatrice di Lecco, Margherita, la porta della speranza è il racconto di un viaggio che coinvolge anche i fratelli di Margherita e suo padre, Michele Baruffaldi, marito di Giuditta Mascheri e già Vicepresidente di Pro RETT Ricerca.
Grazie all'aiuto della professoressa Mariagrazia De Michele, una sua docente ai tempi delle scuole superiori, il "diario" di Giuditta ha preso la forma di un testo pronto ad andare in stampa e quindi a essere distribuito in primis alle altre famiglie Rett; in secundis a tutte quelle persone che sono state coinvolte dalla sua narrazione.

Il libro non si limita a ripercorrere le tappe della vita insieme a Margherita, ma propone un affondo introspettivo capace di andare al di là di un semplice susseguirsi di fatti, quasi volendo proporre al lettore una chiave di lettura più intimistica in grado di toccare l'animo di chiunque si affidi alle sue pagine o alle tante presentazioni pubbliche a cui Michele e Giuditta sono stati chiamati.
Il successo di Margherita, la porta della speranza è testimoniato dalla rassegna stampa che andremo a proporvi tra poco e che terremo aggiornata con il passare dei mesi.
Per sapere come ricevere una copia del libro scriveteci all'indirizzo mail segreteria@prorett.org
- - - - - - - - - - - - -
RASSEGNA STAMPA
Lecco Online – 27 dicembre 2023
malattierare.gov.it – 7 febbraio 2024
{kind=link}
- Margherita, la porta della speranza
- Pasqua 2024 di Pro RETT Ricerca
- Francesca Latella - 21 febbraio 2024
- 16 dicembre a Milano
- Il Natale 2023 di Pro RETT Ricerca
- Azzurra per il 5X1000 2023
- Pro RETT News #07 - marzo 2023
- Pasqua 2023 di Pro RETT Ricerca
- Diventa socio di Pro RETT Ricerca
- Il Natale 2022 di Pro RETT
- Due nuovi progetti nel 2022
- Riconfermato il Consiglio Direttivo
- Il 5x1000 a Pro RETT Ricerca
- Pasqua 2022
- L'Erbolario sostiene la ricerca scientifica
- Il mercatino di Natale 2021
- Dove c'è un bisogno, c'è un Lion
- Secondo incontro online...
- Incontro online...
- Uniti per accelerare...
- Il Natale 2020 di Pro RETT
- Campagna fotografica 2020
- Il Vostro 5x1000 a Pro RETT
- Pasqua 2020 con Pro RETT
- Le Voci di Rett
- 6th European Conference
Pasqua 2024 con Pro RETT Ricerca
Perchè la ricerca non deve fermarsi
12 febbraio 2024
Per la Pasqua 2024 Pro RETT Ricerca sostiene la ricerca di una cura alla sindrome di Rett con le uova di cioccolato artigianali da 350 grammi a fronte di una donazione di 10 euro.
Le nostre famiglie sul territorio distribuiranno le uova di cioccolato fondente e al latte confezionate per noi da un laboratorio artigianale della provincia di Mantova. Vista la delicatezza del cioccolato non abbiamo la possibilità di spedirle al dettaglio, ma scrivete alla nostra Segreteria (segreteria@prorett.org) per qualsiasi informazione o per chiedere se c'è un nostro punto di distribuzione nella vostra zona.

Per restare aggiornati sulle nostre iniziative e sostenere il nostro lavoro vi invitiamo a seguirci sulla pagina Facebook di Pro RETT Ricerca.
Grazie!
- Margherita, la porta della speranza
- Pasqua 2024 di Pro RETT Ricerca
- Francesca Latella - 21 febbraio 2024
- 16 dicembre a Milano
- Il Natale 2023 di Pro RETT Ricerca
- Azzurra per il 5X1000 2023
- Pro RETT News #07 - marzo 2023
- Pasqua 2023 di Pro RETT Ricerca
- Diventa socio di Pro RETT Ricerca
- Il Natale 2022 di Pro RETT
- Due nuovi progetti nel 2022
- Riconfermato il Consiglio Direttivo
- Il 5x1000 a Pro RETT Ricerca
- Pasqua 2022
- L'Erbolario sostiene la ricerca scientifica
- Il mercatino di Natale 2021
- Dove c'è un bisogno, c'è un Lion
- Secondo incontro online...
- Incontro online...
- Uniti per accelerare...
- Il Natale 2020 di Pro RETT
- Campagna fotografica 2020
- Il Vostro 5x1000 a Pro RETT
- Pasqua 2020 con Pro RETT
- Le Voci di Rett
- 6th European Conference
Incontro con la psicoterapeuta dr.ssa Francesca Latella
Mercoledì 21 febbraio 2024, in collegamento remoto su piattaforma Zoom alle ore 21.00

Pro RETT Ricerca ha organizzato un incontro online, per cui è fondamentale iscriversi via mail all'indirizzo segreteria@prorett.org, al fine di presentare un corso di 5 appuntamenti a numero chiuso con la dr.ssa Francesca Latella, pensato e offerto dal Direttivo per venire incontro ad alcune difficoltà comuni a tante famiglie.
Il percorso è rivolto ai caregiver per aiutarli a ricominciare ad amarsi e continuare a prendersi cura di chi si ama senza rinunciare a se stessi.
Per restare aggiornati sulle nostre iniziative e sostenere il nostro lavoro vi invitiamo a seguirci sulla pagina Facebook di Pro RETT Ricerca.
Grazie!
- Margherita, la porta della speranza
- Pasqua 2024 di Pro RETT Ricerca
- Francesca Latella - 21 febbraio 2024
- 16 dicembre a Milano
- Il Natale 2023 di Pro RETT Ricerca
- Azzurra per il 5X1000 2023
- Pro RETT News #07 - marzo 2023
- Pasqua 2023 di Pro RETT Ricerca
- Diventa socio di Pro RETT Ricerca
- Il Natale 2022 di Pro RETT
- Due nuovi progetti nel 2022
- Riconfermato il Consiglio Direttivo
- Il 5x1000 a Pro RETT Ricerca
- Pasqua 2022
- L'Erbolario sostiene la ricerca scientifica
- Il mercatino di Natale 2021
- Dove c'è un bisogno, c'è un Lion
- Secondo incontro online...
- Incontro online...
- Uniti per accelerare...
- Il Natale 2020 di Pro RETT
- Campagna fotografica 2020
- Il Vostro 5x1000 a Pro RETT
- Pasqua 2020 con Pro RETT
- Le Voci di Rett
- 6th European Conference
Sabato 16 dicembre 2023
Festeggia il Natale con Pro RETT Ricerca
01/12/2023
Sabato 16 dicembre 2023 presso gli spazi di Cascina Biblioteca a Milano
Pro RETT Ricerca ha organizzato un momento di condivisione tra i ricercatori che sosteniamo e le famiglie, a cui seguirà un pranzo presso la Trattoria Solidale che da molti anni offre "una ristorazione di qualità con alle spalle progetti di formazione specifica per persone con fragilità psichiche, intellettive e sensoriali, con l’obiettivo di offrire opportunità di lavoro concrete e di inclusione attiva".
Questo il programma della giornata, ricordandovi che la partecipazione è gratuita ma la prenotazione è obbligatoria all'indirizzo segreteria@prorett.org.

Per restare aggiornati sulle nostre iniziative e sostenere il nostro lavoro vi invitiamo a seguirci sulla pagina Facebook di Pro RETT Ricerca.
Grazie!
Puoi effettuare la tua donazione online in totale sicurezza con la tua Carta di Credito o il tuo account PayPal.
Cliccando DONA ADESSO o il Logo PayPal qui sopra verrai reindirizzato al sito PayPal per concludere l'operazione.
- Margherita, la porta della speranza
- Pasqua 2024 di Pro RETT Ricerca
- Francesca Latella - 21 febbraio 2024
- 16 dicembre a Milano
- Il Natale 2023 di Pro RETT Ricerca
- Azzurra per il 5X1000 2023
- Pro RETT News #07 - marzo 2023
- Pasqua 2023 di Pro RETT Ricerca
- Diventa socio di Pro RETT Ricerca
- Il Natale 2022 di Pro RETT
- Due nuovi progetti nel 2022
- Riconfermato il Consiglio Direttivo
- Il 5x1000 a Pro RETT Ricerca
- Pasqua 2022
- L'Erbolario sostiene la ricerca scientifica
- Il mercatino di Natale 2021
- Dove c'è un bisogno, c'è un Lion
- Secondo incontro online...
- Incontro online...
- Uniti per accelerare...
- Il Natale 2020 di Pro RETT
- Campagna fotografica 2020
- Il Vostro 5x1000 a Pro RETT
- Pasqua 2020 con Pro RETT
- Le Voci di Rett
- 6th European Conference
Natale solidale 2023
16 ottobre 2023
Cari amici, si avvicina il Natale e noi siamo già pronti a consegnarvi i nostri regali solidali.

Qui sotto troverete il nostro catalogo solidale (potrete scaricarlo anche qui), rivisto nella grafica e con una selezione di prodotti pensati per garantire a tutti i nostri sostenitori la qualità che contraddistingue le campagne di raccolta fondi di Pro RETT Ricerca.
{kind=link}
Come ogni anno potete contattare la vostra famiglia di riferimento, che raccoglierà il vostro ordine, oppure potete scrivere a segreteria@prorett.org specificando cosa desiderate. In caso di piccole spedizioni è previsto un contributo per le spese di spedizione.
Grazie al vostro aiuto continueremo a sostenere la prof.ssa Landsberger che coordina i progetti del San Raffaele Rett Research Center di Milano e quelli del laboratorio di Biologia Cellulare e Molecolare dell’Università di Milano; il prof. Tongiorgi presso il Laboratorio di Neurobiologia Cellulare dell’Università di Trieste; il dr. Balestra dell‘Università degli Studi di Ferrara e la dr.ssa Antonucci dell‘Università degli Studi di Milano.
Per qualsiasi informazione non esistate a contattarci: vi risponderemo (quasi) subito!

- Margherita, la porta della speranza
- Pasqua 2024 di Pro RETT Ricerca
- Francesca Latella - 21 febbraio 2024
- 16 dicembre a Milano
- Il Natale 2023 di Pro RETT Ricerca
- Azzurra per il 5X1000 2023
- Pro RETT News #07 - marzo 2023
- Pasqua 2023 di Pro RETT Ricerca
- Diventa socio di Pro RETT Ricerca
- Il Natale 2022 di Pro RETT
- Due nuovi progetti nel 2022
- Riconfermato il Consiglio Direttivo
- Il 5x1000 a Pro RETT Ricerca
- Pasqua 2022
- L'Erbolario sostiene la ricerca scientifica
- Il mercatino di Natale 2021
- Dove c'è un bisogno, c'è un Lion
- Secondo incontro online...
- Incontro online...
- Uniti per accelerare...
- Il Natale 2020 di Pro RETT
- Campagna fotografica 2020
- Il Vostro 5x1000 a Pro RETT
- Pasqua 2020 con Pro RETT
- Le Voci di Rett
- 6th European Conference
La tua firma sul 5X1000
Per continuare a sostenere la ricerca di una cura!
5 maggio 2023
Puoi aiutare la ricerca di una cura per Azzurra con una firma sulla dichiarazione dei redditi, specificando il codice fiscale dell'associazione: 93043680201.
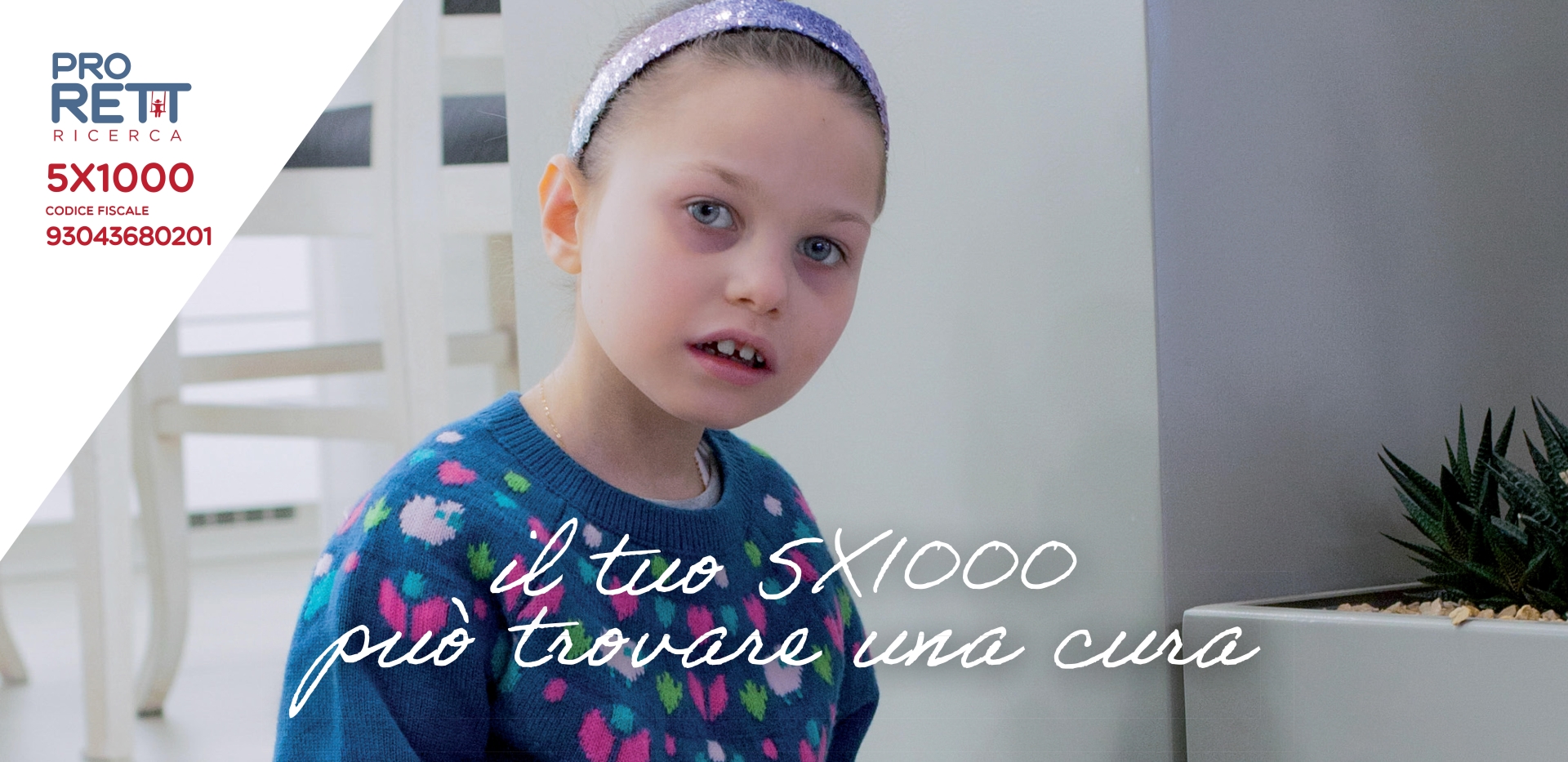
Il volto di Azzurra sarà il testimonial della nostra campagna per il 5X1000 a Pro RETT Ricerca. CON IL SUO AIUTO continueremo a sostenere la ricerca scientifica sulla sindrome di Rett e nelle prossime settimane ti porteremo le testimonianze dei ricercatori che continuano a lavorare grazie al tuo contributo.
Continua a camminare con noi: firma per il tuo 5X1000 a Pro RETT Ricerca sul modello 730, il CU oppure il modello unico nel primo riquadro a "Sostegno degli Enti del Terzo Settore iscritti nel RUNTS, nonché sostegno delle ONLUS iscritte all’anagrafe" e inserisci il nostro codice fiscale: 93043680201.
Scopri le ricerche che stiamo sostenendo sul nuovo numero di Pro RETT News #07 - marzo 2023 (consultandolo qui), mentre se vuoi scaricarlo puoi farlo (cliccando qui).
Puoi consultare tutti i numeri usciti (QUI).
Puoi effettuare la tua donazione online in totale sicurezza con la tua Carta di Credito o il tuo account PayPal.
Cliccando DONA ADESSO o il Logo PayPal qui sopra verrai reindirizzato al sito PayPal per concludere l'operazione.
- Margherita, la porta della speranza
- Pasqua 2024 di Pro RETT Ricerca
- Francesca Latella - 21 febbraio 2024
- 16 dicembre a Milano
- Il Natale 2023 di Pro RETT Ricerca
- Azzurra per il 5X1000 2023
- Pro RETT News #07 - marzo 2023
- Pasqua 2023 di Pro RETT Ricerca
- Diventa socio di Pro RETT Ricerca
- Il Natale 2022 di Pro RETT
- Due nuovi progetti nel 2022
- Riconfermato il Consiglio Direttivo
- Il 5x1000 a Pro RETT Ricerca
- Pasqua 2022
- L'Erbolario sostiene la ricerca scientifica
- Il mercatino di Natale 2021
- Dove c'è un bisogno, c'è un Lion
- Secondo incontro online...
- Incontro online...
- Uniti per accelerare...
- Il Natale 2020 di Pro RETT
- Campagna fotografica 2020
- Il Vostro 5x1000 a Pro RETT
- Pasqua 2020 con Pro RETT
- Le Voci di Rett
- 6th European Conference
È in arrivo il nuovo Pro RETT News di marzo 2023
Un numero interamente dedicato agli aggiornamenti sulle ricerche in corso
22 marzo 2023
È in partenza il nuovo Pro RETT News #07 di marzo 2023, con tutti gli aggiornamenti dai laboratori di ricerca che sosteniamo grazie alle vostre donazioni ma anche dai laboratori di rilevanza internazionale che hanno fatto annunci ricchi di speranza per tutta la comunità Rett.
Pro RETT News #07 arriverà direttamente a casa di tutti i soci e i donatori, gratuitamente e in formato cartaceo, senza che dobbiate fare nulla per riceverlo.

In particolare vi parleremo dei progressi fatti dal progetto di ricerca sulle cellule staminali, con l'avvio di una seconda fase dedicata ai difetti respiratori grazie al pletismografo donato all'Università degli Studi di Milano, dei risultati ottenuti fino a oggi dal Centro Drug Screening finanziato da Pro RETT Ricerca presso l'Università degli Studi di Trieste e dei 2 nuovi progetti di ricerca che abbiamo avviato grazie al bando Spring Seed Grant 2022 di Fondazione Telethon, che ringraziamo per il grande supporto nell'individuazione dei progetti più meritevoli di essere sostenuti economicamente.
Per accedere in anteprima ai contenuti del nuovo numero (cliccate qui), mentre per scaricarlo potete farlo (cliccando qui).
Potete consultare tutti i numeri usciti (QUI).
Puoi effettuare la tua donazione online in totale sicurezza con la tua Carta di Credito o il tuo account PayPal.
Cliccando DONA ADESSO o il Logo PayPal qui sopra verrai reindirizzato al sito PayPal per concludere l'operazione.
- Margherita, la porta della speranza
- Pasqua 2024 di Pro RETT Ricerca
- Francesca Latella - 21 febbraio 2024
- 16 dicembre a Milano
- Il Natale 2023 di Pro RETT Ricerca
- Azzurra per il 5X1000 2023
- Pro RETT News #07 - marzo 2023
- Pasqua 2023 di Pro RETT Ricerca
- Diventa socio di Pro RETT Ricerca
- Il Natale 2022 di Pro RETT
- Due nuovi progetti nel 2022
- Riconfermato il Consiglio Direttivo
- Il 5x1000 a Pro RETT Ricerca
- Pasqua 2022
- L'Erbolario sostiene la ricerca scientifica
- Il mercatino di Natale 2021
- Dove c'è un bisogno, c'è un Lion
- Secondo incontro online...
- Incontro online...
- Uniti per accelerare...
- Il Natale 2020 di Pro RETT
- Campagna fotografica 2020
- Il Vostro 5x1000 a Pro RETT
- Pasqua 2020 con Pro RETT
- Le Voci di Rett
- 6th European Conference
Pasqua 2023 con Pro RETT Ricerca
Perchè la ricerca non deve fermarsi
21 febbraio 2023
Per la Pasqua 2023 Pro RETT Ricerca sostiene la ricerca di una cura alla sindrome di Rett con le uova di cioccolato artigianali da 350 grammi a fronte di una donazione di 10 euro.
Le nostre famiglie sul territorio distribuiranno le uova di cioccolato fondente e al latte confezionate per noi da un laboratorio artigianale della provincia di Mantova. Vista la delicatezza del cioccolato non abbiamo la possibilità di spedirle al dettaglio, ma scrivete alla nostra Segreteria (segreteria@prorett.org) per qualsiasi informazione o per chiedere se c'è un nostro punto di distribuzione nella vostra zona.

Per restare aggiornati sulle nostre iniziative e sostenere il nostro lavoro vi invitiamo a seguirci sulla pagina Facebook di Pro RETT Ricerca.
Grazie!
- Margherita, la porta della speranza
- Pasqua 2024 di Pro RETT Ricerca
- Francesca Latella - 21 febbraio 2024
- 16 dicembre a Milano
- Il Natale 2023 di Pro RETT Ricerca
- Azzurra per il 5X1000 2023
- Pro RETT News #07 - marzo 2023
- Pasqua 2023 di Pro RETT Ricerca
- Diventa socio di Pro RETT Ricerca
- Il Natale 2022 di Pro RETT
- Due nuovi progetti nel 2022
- Riconfermato il Consiglio Direttivo
- Il 5x1000 a Pro RETT Ricerca
- Pasqua 2022
- L'Erbolario sostiene la ricerca scientifica
- Il mercatino di Natale 2021
- Dove c'è un bisogno, c'è un Lion
- Secondo incontro online...
- Incontro online...
- Uniti per accelerare...
- Il Natale 2020 di Pro RETT
- Campagna fotografica 2020
- Il Vostro 5x1000 a Pro RETT
- Pasqua 2020 con Pro RETT
- Le Voci di Rett
- 6th European Conference
Diventa socio di Pro RETT Ricerca
È il primo passo per trovare una cura!
7 febbraio 2023

Dal 2004 il nostro obiettivo è sempre lo stesso: promuovere la ricerca scientifica sulla sindrome di Rett e contribuire attivamente a trovare una cura per le nostre figlie.
Associarsi a Pro RETT Ricerca è:
- il primo passo per sostenere il lavoro dei ricercatori impegnati a studiare questa malattia nei laboratori delle Università italiane;
- il primo passo per contribuire allo sforzo delle nostre famiglie che lavorano incessantemente per raccogliere fondi da destinare ai progetti di ricerca che sosteniamo economicamente;
- è il primo passo per dire alle nostre figlie che ci impegneremo senza risparmiarci affinchè la loro malattia venga studiata e curata.
L'anno che ci siamo lasciati alle spalle è stato un anno molto importante, perchè grazie al contributo di tutti i soci e i donatori siamo riusciti a raddoppiare il nostro impegno per la ricerca.
Ecco un breve riassunto dei progetti che abbiamo sostenuto nel 2022 e che sono ancora in corso di finanziamento:
- uno studio sull'efficacia delle cellule staminali nel trattamento della sindrome di Rett presso l'Università degli Studi di Milano;
- il progetto di Drug Screening per la sindrome di Rett presso l'Università degli Studi di Trieste;
- la caratterizzazione dei difetti astrocitari nella sindrome di Rett presso l'Università di Milano;
- la validazione di due nuovi trattamenti per la sindrome di Rett con un focus sui difetti respiratori della malattia presso l'Università degli Studi di Milano;
- lo studio funzionale per un nuovo possibile approccio farmacologico che agisca sui processi del neurosviluppo presso l’Università degli Studi di Milano;
- la messa a punto di un modello cellulare su cui testare approcci terapeutici basati sull’editing genomico presso l’Università degli Studi di Ferrara;
- lo sviluppo di una nuova piattaforma molecolare per lo screening di farmaci per il trattamento della sindrome di Rett presso il San Raffaele Rett Research Center dell'Ospedale San Raffaele di Milano.
Sempre nel 2022 abbiamo donato 3 strumenti tecnologici per accelerare lo sviluppo dei progetti in corso:
- un pletismografo, che serve a misurare l'efficacia respiratoria dei farmaci su modelli in vivo;
- un vibratomo, che serve a ottenere campioni celebrali di modelli in vivo di grandissima precisione;
- una macchina PCR, che serve a ottenere rapidamente milioni di molecole di DNA a partire da quantità estremamente ridotte dell’acido nucleico.
Per continuare a sostenere questi progetti abbiamo bisogno dell'aiuto di tutti: anche del tuo.
Per associarsi o rinnovare la propria iscrizione è sufficiente un versamento di 50,00 euro (con causale QUOTA SOCIO 2023) intestato a:
Pro Rett Ricerca Onlus
via XXV Aprile 52
46028 Sermide e Felonica (MN)
Il versamento può essere effettuato su uno qualsiasi dei nostri conti:
Conto Corrente Postale n° 55989073
Bonifico bancario Banca Monte Dei Paschi di Siena
IBAN: IT92 G010 3057 9700 0001 0050 057
Bonifico Bancario Banca Intesa San Paolo
IBAN: IT04 I030 6909 6061 0000 0074 468
Bonifico Bancario Banca della Valsassina
IBAN: IT22 X085 1551 3600 0000 0201 216
Online con il tuo conto PayPal con un versameno a info@prorett.org
Puoi effettuare la tua donazione online in totale sicurezza con la tua Carta di Credito o il tuo account PayPal.
Cliccando DONA ADESSO o il Logo PayPal qui sopra verrai reindirizzato al sito PayPal per concludere l'operazione.
- Margherita, la porta della speranza
- Pasqua 2024 di Pro RETT Ricerca
- Francesca Latella - 21 febbraio 2024
- 16 dicembre a Milano
- Il Natale 2023 di Pro RETT Ricerca
- Azzurra per il 5X1000 2023
- Pro RETT News #07 - marzo 2023
- Pasqua 2023 di Pro RETT Ricerca
- Diventa socio di Pro RETT Ricerca
- Il Natale 2022 di Pro RETT
- Due nuovi progetti nel 2022
- Riconfermato il Consiglio Direttivo
- Il 5x1000 a Pro RETT Ricerca
- Pasqua 2022
- L'Erbolario sostiene la ricerca scientifica
- Il mercatino di Natale 2021
- Dove c'è un bisogno, c'è un Lion
- Secondo incontro online...
- Incontro online...
- Uniti per accelerare...
- Il Natale 2020 di Pro RETT
- Campagna fotografica 2020
- Il Vostro 5x1000 a Pro RETT
- Pasqua 2020 con Pro RETT
- Le Voci di Rett
- 6th European Conference
Il Natale solidale 2022 di Pro RETT Ricerca
18 ottobre 2022
Cari amici,
si avvicina il Natale e noi siamo già pronti a consegnarvi i nostri regali solidali.

Come ogni anno potete contattare la vostra famiglia di riferimento, che raccoglierà il vostro ordine, oppure potete scrivere a segreteria@prorett.org specificando cosa desiderate.
Qui sotto troverete il nostro catalogo solidale (potrete scaricarlo anche qui), rivisto nella grafica e con una selezione di prodotti pensati per garantire a tutti i nostri sostenitori la qualità che contraddistingue le campagne di raccolta fondi di Pro RETT Ricerca.
Grazie al vostro aiuto continueremo a sostenere il Laboratorio del San Raffaele Rett Research Center di Milano, il Laboratorio di Biologia Cellulare e Molecolare Applicata a Patologie del Neurosviluppo dell’Università degli Studi di Milano, il Laboratorio di Neurobiologia Cellulare e dello Sviluppo presso il Dipartimento di Scienze della Vita dell’Università di Trieste.
Per qualsiasi informazione non esistate a contattarci: vi risponderemo (quasi) subito!

- Margherita, la porta della speranza
- Pasqua 2024 di Pro RETT Ricerca
- Francesca Latella - 21 febbraio 2024
- 16 dicembre a Milano
- Il Natale 2023 di Pro RETT Ricerca
- Azzurra per il 5X1000 2023
- Pro RETT News #07 - marzo 2023
- Pasqua 2023 di Pro RETT Ricerca
- Diventa socio di Pro RETT Ricerca
- Il Natale 2022 di Pro RETT
- Due nuovi progetti nel 2022
- Riconfermato il Consiglio Direttivo
- Il 5x1000 a Pro RETT Ricerca
- Pasqua 2022
- L'Erbolario sostiene la ricerca scientifica
- Il mercatino di Natale 2021
- Dove c'è un bisogno, c'è un Lion
- Secondo incontro online...
- Incontro online...
- Uniti per accelerare...
- Il Natale 2020 di Pro RETT
- Campagna fotografica 2020
- Il Vostro 5x1000 a Pro RETT
- Pasqua 2020 con Pro RETT
- Le Voci di Rett
- 6th European Conference
Due nuovi progetti di ricerca!
Pro RETT Ricerca ha finanziato due nuovi progetti di ricerca promossi dal bando Spring Seed Grant 2022 di Fondazione Telethon
19 settembre 2022
Cari amici,
è con enorme piacere che finalmente possiamo rendervi partecipi di un'iniziativa che ha coinvolto la nostra associazione per diversi mesi e che ha portato al finanziamento di due nuovi progetti di ricerca sulla sindrome di Rett.

Grazie al bando "Spring Seed Grant 2022" di Fondazione Telethon sosterremo economicamente il lavoro della dr.ssa Flavia Antonucci, presso l’Università degli Studi di Milano, che realizzerà uno studio funzionale per un nuovo possibile approccio farmacologico che agisca sui processi del neurosviluppo, e il dr. Dario Balestra dell’Università degli Studi di Ferrara, che si focalizzerà sulla messa a punto di un modello cellulare su cui testare approcci terapeutici basati sull’editing genomico, per correggere alcune delle mutazioni più comuni associate alla sindrome di Rett.
Trovate il video integrale della presentazione anche sulla nostra pagina Facebook e nei prossimi mesi non mancheremo di aggiornarvi sui progressi dei due singoli progetti di ricerca, quindi seguiteci!
Per ora vi lasciamo al comunicato stampa di Fondazione Telethon che spiega l'importanza dei bandi "Seed Grant" per le associazioni di pazienti e di come sia possibile "fare la differenza" lavorando insieme.
- - - - - - - - - - - - - - - - -
DA FONDAZIONE TELETHON E ASSOCIAZIONI DI PAZIENTI
500 MILA EURO PER 10 PROGETTI “SEED”
SU 6 MALATTIE GENETICHE RARE
Giunta alla sua quarta edizione, l’iniziativa Seed Grant offre alle associazioni di pazienti un supporto per investire al meglio i propri fondi, selezionando i progetti di ricerca più meritevoli sulle patologie di interesse
9 settembre 2022 – Annunciati da Fondazione Telethon i vincitori del bando “Spring Seed Grant” 2022: sono dieci i progetti di ricerca finanziati per lo studio di sei malattie genetiche rare, per un totale di 500 mila euro. Avviata per la prima volta dalla Fondazione nel 2019, l’iniziativa ha l’obiettivo di aiutare le associazioni di pazienti con malattie genetiche rare a investire in modo mirato i propri fondi, attraverso la selezione dei migliori progetti di ricerca sulle proprie patologie di interesse. In questa quarta edizione del bando sono stati finanziati progetti sulla sindrome di Phelan Mc-Dermid, la malattia di Fabry, la sindrome di Rett, la sindrome emolitico-uremica (Seu) atipica, la malattia di Lafora e la sindrome del neurosviluppo dovuta a mutazioni del gene CAMK2b.
Sette invece le associazioni coinvolte: Associazione Italiana per la Sindrome di Phelan-McDermid, Associazione Uniphelan, Associazione Italiana Anderson-Fabry, Pro RETT Ricerca - Associazione per la ricerca sulla sindrome di Rett ONLUS, Progetto Alice Onlus - Associazione per la lotta alla SEU, Associazione Tempo Zero, Associazione UNICI.
Come indica la parola seed, ogni finanziamento rappresenta un “seme” da gettare per iniziare un percorso di ricerca su un tema scarsamente studiato, come avviene spesso nell’ambito delle malattie genetiche rare. Ogni progetto è stato valutato da una commissione scientifica, costituita ad hoc e formata da ricercatori di fama internazionale esperti della patologia, che ha individuato quali fossero meritevoli di finanziamento, per poi presentarli alle rispettive associazioni, perché si giungesse a una decisione finale.
Ad oggi sono complessivamente 37 i "seed grant" finanziati, per studiare 20 malattie genetiche rare, per oltre 1,8 milioni di euro di investimento totale. Ventuno le associazioni coinvolte, oltre a una piccola fondazione privata, ma anche Fondazione Telethon stessa, che ha voluto fare la sua parte finanziando 8 progetti tra quelli selezionati come meritevoli e presentati da ricercatori dei propri Istituti di Milano e Pozzuoli.
I progetti finanziati nella quarta edizione
Due i progetti finanziati sulla sindrome di Phelan-McDermid, che a partire dai 2-3 anni compromette sensibilmente lo sviluppo motorio e intellettivo. Il progetto di Letizia Allegra Mascaro del Consiglio Nazionale delle Ricerche (CNR) di Firenze studierà come monitorare e modulare l’attività delle aree della corteccia cerebrale coinvolte nell’elaborazione degli stimoli sensoriali. L’auspicio è che grazie alla comprensione dei processi neurobiologici alla base dei sintomi della malattia si potranno migliorare i difetti neurologici tipici di questi pazienti. All’Università Vita Salute San Raffaele di Milano, invece, Luca Colnaghi si concentrerà su alcune particolari mutazioni di SHANK3, il principale gene in cui ricadono le mutazioni associate alla sindrome. In particolare, saranno caratterizzate alcune regioni della proteina codificata dal gene SHANK3 dalla funzione ancora poco nota e l’interazione con altre proteine.
Sempre due i progetti dedicati allo studio della malattia di Anderson-Fabry, caratterizzata dall’accumulo nelle cellule di un particolare gruppo di zuccheri e che può portare, nei casi più gravi, a insufficienza renale, infarto o ictus. Il progetto di Maria Antonietta De Matteis dell’Istituto Telethon di genetica e medicina (Tigem) di Pozzuoli si concentrerà sullo sviluppo di saggi cellulari per indagare alcune varianti ancora poco studiate del gene GLA, che codifica per l’enzima carente nella malattia e responsabile della degradazione di quei particolari zuccheri. Paola Sacerdote dell’Università degli Studi di Milano andrà alla ricerca di nuove possibili strategie farmacologiche per alleviare il dolore e i disturbi dell'umore tipici di questa malattia.
Due progetti saranno poi dedicati alla sindrome di Rett, malattia neurologica che colpisce prevalentemente il sesso femminile e rappresenta una delle più comuni cause di disabilità grave. Flavia Antonucci dell’Università degli Studi di Milano realizzerà uno studio funzionale per un nuovo possibile approccio farmacologico che agisca sui processi del neurosviluppo. Dario Balestra dell’Università degli Studi di Ferrara si focalizzerà sulla messa a punto di un modello cellulare su cui testare approcci terapeutici basati sull’editing genomico, per correggere alcune delle mutazioni più comuni associate alla sindrome di Rett.
Il progetto dedicato alla sindrome emolitico-uremica (Seu) atipica, rara malattia caratterizzata da anemia emolitica, diminuzione del numero di piastrine e danno renale, sarà quello di Ilaria Bestetti della Fondazione IRCCS Ca' Granda - Ospedale Maggiore Policlinico di Milano: con il suo gruppo di ricerca si focalizzerà sul ruolo di varianti epigenetiche. Queste alterazioni non sono cioè a livello della sequenza del Dna ma, per esempio, nelle modifiche chimiche che ne regolano l’accesso o il ripiegamento nello spazio e che tipicamente risentono di influssi ambientali. Questo studio valuterà un approccio genomico mirato a identificare potenziali differenze epigenetiche tra pazienti con SEU atipica, per spiegare i fattori che causano lo sviluppo di autoimmunità.
Altri due progetti riguarderanno la malattia di Lafora (LD), una grave forma di epilessia mioclonica progressiva ereditaria, caratterizzata da un significativo e rapido deterioramento cognitivo e motorio. Paolo Prontera dell’Azienda Ospedaliera di Perugia studierà se particolari sequenze di RNA non codificanti sintetici, detti SINEUP, siano in grado in vitro di degradare gli accumuli tossici tipici della malattia, i cosiddetti corpi di Lafora. Stefania Della Vecchia, ricercatrice presso la Fondazione IRCCS Stella Maris di Pisa, utilizzerà un modello di pesce zebra, già messo a punto in un suo studio precedente, per caratterizzare la microglia, un particolare tipo di cellule di supporto presenti nel sistema nervoso, e i meccanismi cellulari coinvolti nella neuro-infiammazione, tipica della malattia.
Infine, il progetto dedicato alle mutazioni del gene CAMK2b, associate a un raro disturbo che ricade nello spettro autistico, vedrà Claudia Compagnucci dell’Ospedale Pediatrico Bambino Gesù di Roma impegnata nella generazione e caratterizzazione funzionale delle cellule staminali pluripotenti indotte (IPS), derivate cioè da cellule dei pazienti, per comprendere i meccanismi patologici associati a queste particolari mutazioni.
“Il bando “seed grant”, arrivato ormai alla sua quarta edizione, si è consolidato negli anni, con sempre più associazioni di pazienti che scelgono di investire i propri fondi in ricerca scientifica – dichiara Francesca Pasinelli, Direttore Generale di Fondazione Telethon – Fondazione Telethon mette a disposizione le proprie competenze lanciando il bando, creando le commissioni internazionali di esperti scelti ad hoc, che valutino la qualità scientifica e il potenziale impatto sui pazienti dei progetti raccolti, e presentando alle associazioni quelli ritenuti meritevoli di finanziamento. A questo punto, sono le associazioni stesse a scegliere quale ritengono più utili o di interesse per i propri pazienti. Il numero sempre più alto di associazioni di pazienti che si rivolge a noi per il bando “Seed Grant” è motivo di grande soddisfazione, perché dimostra la fiducia che esse ripongono nel nostro metodo di selezione della ricerca e impegno per le malattie genetiche rare, che troppo spesso ricevono scarsa attenzione e risorse”.
“Lavorare accanto alle famiglie e alle associazioni è fondamentale per noi, perché siamo consci di quali siano le difficoltà quotidiane di chi convive con una malattia genetica rara - dichiara Alessandra Camerini, Responsabile Relazioni con i Pazienti e le Associazioni – Questa attività, come diverse altre, è un’ulteriore opportunità per unire le forze e permetterci di mettere a loro disposizione la nostra esperienza trentennale per dare un supporto concreto in un ambito complesso, come quello dell’organizzazione di un bando di ricerca, ed investire in progetti meritevoli, non disperdendo i fondi raccolti con tanto sforzo”.
Per ulteriori informazioni e dettagli sui ricercatori, le malattie e i progetti di ricerca finanziati, visita il sito www.telethon.it
Fondazione Telethon ETS
Fondazione Telethon ETS è una delle principali charity biomediche italiane, nata nel 1990 per iniziativa di un gruppo di pazienti affetti da distrofia muscolare. La sua missione è di arrivare alla cura delle malattie genetiche rare grazie a una ricerca scientifica di eccellenza, selezionata secondo le migliori prassi condivise a livello internazionale. Attraverso un metodo unico nel panorama italiano, segue l’intera “filiera della ricerca” occupandosi della raccolta fondi, della selezione e del finanziamento dei progetti e dell’attività stessa di ricerca portata avanti nei centri e nei laboratori della Fondazione. Telethon inoltre sviluppa collaborazioni con istituzioni sanitarie pubbliche e industrie farmaceutiche per tradurre i risultati della ricerca in terapie accessibili ai pazienti. Dalla sua fondazione ha investito in ricerca oltre 592,5 milioni di euro, ha finanziato 2.720 progetti con 1.630 ricercatori coinvolti e più di 580 malattie studiate. Ad oggi grazie a Fondazione Telethon è stata resa disponibile la prima terapia genica con cellule staminali al mondo, nata grazie alla collaborazione con l’industria farmaceutica. Strimvelis, questo il nome commerciale della terapia, è destinata al trattamento dell’ADA-SCID, una grave immunodeficienza che compromette le difese dell’organismo fin dalla nascita. Un’altra terapia genica frutto della ricerca Telethon resa disponibile è quella per una grave malattia neurodegenerativa, la leucodistrofia metacromatica, dal nome commerciale di Libmeldy. Questo approccio terapeutico è in fase avanzata di sperimentazione clinica per un’altra immunodeficienza, la sindrome di Wiskott-Aldrich. Altre malattie su cui la terapia genica messa a punto dai ricercatori Telethon è stata valutata nei pazienti sono la beta talassemia e due malattie metaboliche dell’infanzia, la mucopolisaccaridosi di tipo 6 e di tipo 1. Inoltre, all’interno degli istituti Telethon è in fase avanzata di studio o di sviluppo una strategia terapeutica mirata anche per altre malattie genetiche, come per esempio l’emofilia o diversi difetti ereditari della vista. Parallelamente, continua in tutti i laboratori finanziati da Telethon lo studio dei meccanismi di base e di potenziali approcci terapeutici per patologie ancora senza risposta.
Per maggiori informazioni:
Ufficio stampa Telethon
HAVAS PR Milan
Thomas Balanzoni – thomas.balanzoni@havaspr.com – tel. 02 85457047 - 346 3204520
Giovanna Giacalone – giovanna.giacalone@havaspr.com – tel. 02 85457053 - 366 6123607
Puoi effettuare la tua donazione online in totale sicurezza con la tua Carta di Credito o il tuo account PayPal.
Cliccando DONA ADESSO o il Logo PayPal qui sopra verrai reindirizzato al sito PayPal per concludere l'operazione.
- Margherita, la porta della speranza
- Pasqua 2024 di Pro RETT Ricerca
- Francesca Latella - 21 febbraio 2024
- 16 dicembre a Milano
- Il Natale 2023 di Pro RETT Ricerca
- Azzurra per il 5X1000 2023
- Pro RETT News #07 - marzo 2023
- Pasqua 2023 di Pro RETT Ricerca
- Diventa socio di Pro RETT Ricerca
- Il Natale 2022 di Pro RETT
- Due nuovi progetti nel 2022
- Riconfermato il Consiglio Direttivo
- Il 5x1000 a Pro RETT Ricerca
- Pasqua 2022
- L'Erbolario sostiene la ricerca scientifica
- Il mercatino di Natale 2021
- Dove c'è un bisogno, c'è un Lion
- Secondo incontro online...
- Incontro online...
- Uniti per accelerare...
- Il Natale 2020 di Pro RETT
- Campagna fotografica 2020
- Il Vostro 5x1000 a Pro RETT
- Pasqua 2020 con Pro RETT
- Le Voci di Rett
- 6th European Conference
L'Assemblea dei soci ha rieletto il Consiglio Direttivo
19 giugno 2022
In occasione dell'Assemblea Ordinaria del 18 giugno 2022, i soci hanno riconfermato il Consiglio Direttivo dell'associazione, dando il benvenuto a un nuovo Consigliere: Andrea Bianco*.

Salvatore Franzé è stato rieletto Presidente, così come Michele Baruffaldi con la carica di Vicepresidente. Sono stati rieletti anche i Consiglieri Rita Bernardelli, Giovanna Lembo (non presente in foto), Orietta Mariotti e Alessandro Sissa.
Facciamo loro un grande "in bocca al lupo" con la certezza che sapranno continuare a lavorare con genuinità e determinazione alla ricerca di una cura per la sindrome di Rett... perchè una cura è possibile e solo attraverso la ricerca scientifica riusciremo a trovarla!
*Andrea Bianco ha rassegnato le proprie dimissioni dalla carica di Consigliere in data 7 novembre 2023 e da quella data non è più parte del Consiglio Direttivo dell'associazione.
Puoi effettuare la tua donazione online in totale sicurezza con la tua Carta di Credito o il tuo account PayPal.
Cliccando DONA ADESSO o il Logo PayPal qui sopra verrai reindirizzato al sito PayPal per concludere l'operazione.
- Margherita, la porta della speranza
- Pasqua 2024 di Pro RETT Ricerca
- Francesca Latella - 21 febbraio 2024
- 16 dicembre a Milano
- Il Natale 2023 di Pro RETT Ricerca
- Azzurra per il 5X1000 2023
- Pro RETT News #07 - marzo 2023
- Pasqua 2023 di Pro RETT Ricerca
- Diventa socio di Pro RETT Ricerca
- Il Natale 2022 di Pro RETT
- Due nuovi progetti nel 2022
- Riconfermato il Consiglio Direttivo
- Il 5x1000 a Pro RETT Ricerca
- Pasqua 2022
- L'Erbolario sostiene la ricerca scientifica
- Il mercatino di Natale 2021
- Dove c'è un bisogno, c'è un Lion
- Secondo incontro online...
- Incontro online...
- Uniti per accelerare...
- Il Natale 2020 di Pro RETT
- Campagna fotografica 2020
- Il Vostro 5x1000 a Pro RETT
- Pasqua 2020 con Pro RETT
- Le Voci di Rett
- 6th European Conference
Il vostro 5x1000 può trovare una cura
31 Maggio 2021
In questi mesi, come milioni di italiani, sarete chiamati a depositare la dichiarazione dei redditi percepiti nel corso del 2021. In quell'occasione avrete la possibilità di scegliere a chi destinare il vostro 5x1000.

Il 5x1000 è una quota della vostra imposta IRPEF, quindi non vi verranno chiesti ulteriori oneri, che lo Stato ripartisce per dare sostegno a enti che svolgono attività socialmente rilevanti, ossia che lavorano per l'interesse proprio della pluralità e della collettività. Il cittadino contribuente può effettuare una scelta precisa, come per l’8 e il 2x1000, mentre la quota di coloro che non indicano un codice fiscale specifico viene ripartita proporzionalmente a seconda delle scelte sottoscritte dagli altri.
Noi di Pro RETT Ricerca siamo genitori volontari, non percepiamo stipendio ma lavoriamo, tutti i giorni, per trovare i fondi necessari a finanziare la ricerca scientifica sulla sindrome di Rett nella speranza di arrivare presto a una cura per le nostre figlie.
Se state leggendo queste pagine significa che non abbiamo bisogno di spiegarvi cosa comporti essere ammalati della sindrome di Rett. Siamo una piccola onlus, mandiamo poche comunicazioni dirette e solo a contatti selezionati, quindi sappiamo per certo che sapete già tutto ciò di cui avete bisogno per capire quanto sia importante, per noi e le nostre figlie, che scegliate Pro RETT Ricerca come destinataria di quelle tasse che avete già pagato.
Come fare?
Cercate nel CUD rilasciato dal datore di lavoro o dall’INPS la pagina dedicata alla scelte, con un campo per la firma e uno per il Codice Fiscale della realtà che volete sostenere. Nella sezione 5x1000, in alto a sinistra, mettete la vostra firma nella casella "Sostegno degli Enti del Terzo settore [...] e delle Onlus iscritte all'anagrafe" e inserite il nostro Codice Fiscale:
93043680201
Nel caso non foste chiamati a compilare la dichiarazione dei redditi avrete comunque la possibilità di destinare il vostro 5x1000 a Pro RETT Ricerca consegnando quella pagina, debitamente compilata e in busta chiusa con l’indicazione del vostro codice fiscale, presso un Ufficio Postale o un CAF abilitato alla trasmissione telematica (ma ormai lo sono quasi tutti).

Oggi, però, vi chiediamo uno sforzo maggiore. Molte persone scelgono a chi destinare il 5x1000 solo per poter chiudere la compilazione della dichiarazione dei redditi alla svelta e non pensarci più. Ecco: portate la nostra voce tra i vostri colleghi, fate giungere la nostra storia ai vostri familiari e agli amici che incontrate dopo il lavoro. Raccontate loro di quello che facciamo; spiegategli che quel 5x1000 “donato” a Pro RETT Ricerca viene utilizzato per pagare lo stipendio dei ricercatori che studiano questa malattia rara e che altrimenti non potrebbero farlo in Italia, per rinnovare la strumentazione e acquistare materiali di consumo nei laboratori dell'Università degli Studi di Milano e presso il San Raffaele Rett Research Center.
Aiutateci a spargere la voce; aiutateci a farlo sapere a tutti.
---
Ringraziamo la fotografa Cristina Corsi per lo scatto alla nostra Beatrice. Cristina lavora insieme al fotografo Antonio Lorenzini per un progetto sulla sindrome di Rett di cui vi parleremo presto.
Puoi effettuare la tua donazione online in totale sicurezza con la tua Carta di Credito o il tuo account PayPal.
Cliccando DONA ADESSO o il Logo PayPal qui sopra verrai reindirizzato al sito PayPal per concludere l'operazione.
- Margherita, la porta della speranza
- Pasqua 2024 di Pro RETT Ricerca
- Francesca Latella - 21 febbraio 2024
- 16 dicembre a Milano
- Il Natale 2023 di Pro RETT Ricerca
- Azzurra per il 5X1000 2023
- Pro RETT News #07 - marzo 2023
- Pasqua 2023 di Pro RETT Ricerca
- Diventa socio di Pro RETT Ricerca
- Il Natale 2022 di Pro RETT
- Due nuovi progetti nel 2022
- Riconfermato il Consiglio Direttivo
- Il 5x1000 a Pro RETT Ricerca
- Pasqua 2022
- L'Erbolario sostiene la ricerca scientifica
- Il mercatino di Natale 2021
- Dove c'è un bisogno, c'è un Lion
- Secondo incontro online...
- Incontro online...
- Uniti per accelerare...
- Il Natale 2020 di Pro RETT
- Campagna fotografica 2020
- Il Vostro 5x1000 a Pro RETT
- Pasqua 2020 con Pro RETT
- Le Voci di Rett
- 6th European Conference
La Pasqua 2022 delle famiglie di Pro RETT Ricerca
28 marzo 2022
Anche quest'anno Pro RETT Ricerca sostiene la ricerca di una cura alla sindrome di Rett con la campagna di raccolta fondi pasquale.
Le nostre famiglie sul territorio distribuiranno le uova di cioccolato fondente e al latte confezionate per noi da un laboratorio artigianale della provincia di Mantova. Vista la delicatezza del cioccolato non abbiamo la possibilità di spedirle al dettaglio, ma scrivete alla nostra Segreteria (segreteria@prorett.org) per qualsiasi informazione o per chiedere se c'è un nostro punto di distribuzione nella vostra zona.

Per restare aggiornati sulle nostre iniziative e sostenere il nostro lavoro vi invitiamo a seguirci sulla pagina Facebook di Pro RETT Ricerca.
Grazie!
- Margherita, la porta della speranza
- Pasqua 2024 di Pro RETT Ricerca
- Francesca Latella - 21 febbraio 2024
- 16 dicembre a Milano
- Il Natale 2023 di Pro RETT Ricerca
- Azzurra per il 5X1000 2023
- Pro RETT News #07 - marzo 2023
- Pasqua 2023 di Pro RETT Ricerca
- Diventa socio di Pro RETT Ricerca
- Il Natale 2022 di Pro RETT
- Due nuovi progetti nel 2022
- Riconfermato il Consiglio Direttivo
- Il 5x1000 a Pro RETT Ricerca
- Pasqua 2022
- L'Erbolario sostiene la ricerca scientifica
- Il mercatino di Natale 2021
- Dove c'è un bisogno, c'è un Lion
- Secondo incontro online...
- Incontro online...
- Uniti per accelerare...
- Il Natale 2020 di Pro RETT
- Campagna fotografica 2020
- Il Vostro 5x1000 a Pro RETT
- Pasqua 2020 con Pro RETT
- Le Voci di Rett
- 6th European Conference
L'Erbolario sostiene Pro RETT Ricerca
PERCHÉ LA CURA È POSSIBILE
28 febbraio 2022
L’Erbolario, marchio lodigiano di cosmesi di derivazione vegetale, in occasione della Giornata Mondiale delle Malattie Rare, lancia una raccolta fondi online di 24 ore a sostegno di Pro RETT Ricerca, associazione italiana che sostiene e finanzia la ricerca scientifica nazionale e internazionale sulla sindrome di Rett.
Una giornata di shopping solidale, dunque, che consentirà a L’Erbolario di dare un sostegno importante al progetto coordinato dalla prof.ssa Nicoletta Landsberger nel suo Laboratorio di Biologia Cellulare e Molecolare Applicata a Patologie del Neurosviluppo presso l’Università degli Studi di Milano, sull’efficacia terapeutica del trapianto di cellule staminali per i trattamenti della sindrome di Rett.

Durante tutta la giornata di lunedì 28 febbraio sarà possibile supportare l’iniziativa acquistando qualunque prodotto L’Erbolario – viso, corpo, capelli e casa - direttamente sul sito e-commerce del marchio cosmetico.
Cosa aspettate?!
- Margherita, la porta della speranza
- Pasqua 2024 di Pro RETT Ricerca
- Francesca Latella - 21 febbraio 2024
- 16 dicembre a Milano
- Il Natale 2023 di Pro RETT Ricerca
- Azzurra per il 5X1000 2023
- Pro RETT News #07 - marzo 2023
- Pasqua 2023 di Pro RETT Ricerca
- Diventa socio di Pro RETT Ricerca
- Il Natale 2022 di Pro RETT
- Due nuovi progetti nel 2022
- Riconfermato il Consiglio Direttivo
- Il 5x1000 a Pro RETT Ricerca
- Pasqua 2022
- L'Erbolario sostiene la ricerca scientifica
- Il mercatino di Natale 2021
- Dove c'è un bisogno, c'è un Lion
- Secondo incontro online...
- Incontro online...
- Uniti per accelerare...
- Il Natale 2020 di Pro RETT
- Campagna fotografica 2020
- Il Vostro 5x1000 a Pro RETT
- Pasqua 2020 con Pro RETT
- Le Voci di Rett
- 6th European Conference
Sostieni la ricerca anche a Natale!
25 novembre 2021
Il Natale si avvicina e anche quest'anno Pro RETT Ricerca propone i suoi prodotti solidali per sostenere la ricerca di una cura per la sindrome di Rett durante le festività.
Sul nostro mercatino online potrai scoprire i panettoni artigianali classici o al cioccolato, i nocciolati della Tuscia, le confezioni regalo e i pandori realizzati apposta per noi con grande attenzione alle materie prime e al confezionamento.

Per qualsiasi domanda o richiesta particolare scrivi subito alla nostra Segreteria (segreteria@prorett.org) e ti risponderemo in giornata, oppure contattaci tramite il format automatico cliccando qui.
Ti aspettiamo!
- Margherita, la porta della speranza
- Pasqua 2024 di Pro RETT Ricerca
- Francesca Latella - 21 febbraio 2024
- 16 dicembre a Milano
- Il Natale 2023 di Pro RETT Ricerca
- Azzurra per il 5X1000 2023
- Pro RETT News #07 - marzo 2023
- Pasqua 2023 di Pro RETT Ricerca
- Diventa socio di Pro RETT Ricerca
- Il Natale 2022 di Pro RETT
- Due nuovi progetti nel 2022
- Riconfermato il Consiglio Direttivo
- Il 5x1000 a Pro RETT Ricerca
- Pasqua 2022
- L'Erbolario sostiene la ricerca scientifica
- Il mercatino di Natale 2021
- Dove c'è un bisogno, c'è un Lion
- Secondo incontro online...
- Incontro online...
- Uniti per accelerare...
- Il Natale 2020 di Pro RETT
- Campagna fotografica 2020
- Il Vostro 5x1000 a Pro RETT
- Pasqua 2020 con Pro RETT
- Le Voci di Rett
- 6th European Conference
25.000 euro alla ricerca grazie al Lions Club Viadana Oglio Po
30 Settembre 2021

Sabato 25 settembre, presso il Palazzo d'Arco di Mantova, gli amici del Lions Club Viadana Oglio Po hanno organizzato un evento per consegnare alla prof.ssa Nicoletta Landsberger i fondi per sostenere una borsa di ricerca sulla terapia cellulare.

L'obiettivo, raggiunto con successo e determinazione, è frutto di un lavoro iniziato nel febbraio 2018, quando il Lions Club Viadana Oglio Po si è avvicinato alla causa di Pro RETT Ricerca – grazie all'intervento di Rodolfo Terzi e Sara Malacarne, mamma della nostra Sofia - organizzando alcuni appuntamenti di sensibilizzazione per promuovere una raccolta fondi al fine di trovare una cura alla sindrome di Rett.
L’idea era quella di costituire un service capace di sostenere il costo di un ricercatore impegnato in progetto di ricerca sulla terapia cellulare; un progetto ambizioso che permettesse di fare un ulteriore passo in avanti in direzione della cura.
Dopo i primi contatti informali e una riunione allargata a cui erano presenti il Presidente di Pro RETT Ricerca, Salvatore Franzè, e la prof.ssa Nicoletta Landsberger, responsabile dei laboratori che studiano la Rett presso l'Università degli Studi di Milano e il Rett Research Center dell'Ospedale San Raffaele di Milano, il 23 marzo 2019 ha preso vita un convegno organizzato dal Lions Club Viadana Oglio Po presso gli spazi museali del Palazzo Ducale di Mantova, a cui hanno presenziato autorità locali, ricercatori, famiglie e le principali cariche istituzionali territoriali dei Lions Clubs International.

Con lo scopo di presentare il progetto di ricerca selezionato per la sovvenzione e gettare le basi per consolidare il service “Adotta un ricercatore” su scala distrettuale, sono state invitate sul palco la dr.ssa Angelisa Frasca dell’Università degli Studi di Milano e la dr.ssa Sara Carli dell’Ospedale San Raffaele, che hanno illustrato le premesse del loro studio sull’efficacia terapeutica del trapianto di cellule staminali per il trattamento della sindrome di Rett.
Sono intervenuti, a vario titolo, anche gli esponenti delle sedi locali dei Lions, ciascuno portando un contributo specifico sia in merito all’urgenza di sostenere “come un sol uomo” il lungo percorso per trovare una cura a questa malattia, sia in relazione all’importanza di sostenere l’eccellenza italiana in campo scientifico laddove le istituzioni non riescono ad arrivare per cause di forza maggiore.
Tra questi vogliamo ringraziare Primo Barzoni, l'allora Presidente della sede di Viadana Oglio Po; Alessandro Colombo, Presidente della sede di Mantova Host e Maria Federica Pasotti, Governatore del multidistretto dei Lions che raggruppa le province di Bergamo, Brescia e Mantova.
Da quel giorno, il Lions Club Viadana Oglio Po non ha mai smesso di lavorare per raggiungere il suo obiettivo:
Il 7 novembre del 2019 ha organizzato uno spettacolo teatrale, intitolato "Du scarpulein curius”, portato al Cinema Teatro Vittoria di Viadana dalla compagnia I Maltaià.

Il 2 febbraio del 2020 si è tenuta la terza edizione della “Cross in Garzaia”, una corsa campestre certificata dagli enti che si occupano di atletica leggera e dei suoi professionisti. Atletica Viadana, in collaborazione con la Fidal Mantova, ha aderito al progetto "Adotta un ricercatore” del Lions Club Viadana Oglio Po per sostenere la ricerca sulla sindrome di Rett, devolvendo al service una parte della quota di iscrizione.

Sempre nel 2020, mentre tutto il mondo era bloccato dalla pandemia causata dal Covid-19, il Lions Club Viadana Oglio Po ha organizzato una campagna di raccolta fondi online (qui) organizzata da Luca Salvagni, attuale presidente del Club, su piattaforma GoFundMe.
Dal 3 al 5 settembre 2021 i Lions hanno quindi partecipato alla fiera patrocinata dal Comune di Viadana in cui era presente un loro stand, con mamma Sara e sua figlia Sofia, attraverso il quale hanno raccolto gli ultimi fondi necessari a raggiungere l’obiettivo.

Arriviamo così a sabato 25 settembre 2001, quando nella splendida cornice di Palazzo d’Arco a Mantova è avvenuta la simbolica consegna dell’assegno di 25 mila euro alla presenza della prof.ssa Nicoletta Landsberger e di Rita Bernardelli, fondatrice e membro del Direttivo di Pro RETT Ricerca, che ha sintetizzato i 15 anni di storia di Pro RETT Ricerca al fianco delle nostre figlie malate.
All'incontro non potevano mancare i principali attori di questa iniziativa, in primis Luca Salvagni - attuale presidente del Lions Club Viadana Oglio Po – a cui vanno i nostri più sinceri ringraziamenti. Senza la sua determinazione, il suo impegno costante e la ferrea volontà di fare la differenza, oggi non stareste leggendo questo articolo.

C'erano anche Ivo Benedetti, Governatore del Distretto Lions 108Ib2, che ha ricordato il motto “Dove c’è un bisogno, c’è un Lion”; Fulvio Venturi, Presidente della Fondazione Bruno Bnà, che ha riferito del sostegno fornito al service tramite i fondi del 5X1000; Christian Manfredi, Presidente di Circoscrizione Mantova, che ha sottolineato quanto il successo del service sia dipeso dallo sforzo congiunto di tutti (Distretto, Fondazione e Clubs) "nella convinzione che solo facendo rete si attua il principio di sussidiarietà per il conseguimento del bene comune”.
Al termine della cerimonia, il Conservatore della Fondazione d’Arco Italo Scaietta ha guidato i presenti alla visita delle magnifiche sale di Palazzo D'Arco, concludendo quella che si preannuncia essere solo la "prima fase" di una collaborazione che potrebbe continuare nel tempo.
Vogliamo quindi ringraziare nuovamente tutti gli amici dei Lions Club del territorio perché in un periodo come questo, quando il rischio di sentirsi da soli e isolati è sempre alle porte, la loro vicinanza e il loro supporto concreto sono stati inestimabili.

Gli ultimi "grazie" vanno a Rodolfo Terzi, Sara Malacarne, la dolce Sofia e nonno Sergio (fotografo professionista che ha seguito tutte le iniziative pubbliche dei Lions), che sul territorio di Viadana sono stati i pensieri, la voce e le mani di tutte le famiglie Rett della nostra associazione ma non solo, perché quando troveremo una cura la troveremo per tutte le bambine o le ragazze malate nel mondo.
- Margherita, la porta della speranza
- Pasqua 2024 di Pro RETT Ricerca
- Francesca Latella - 21 febbraio 2024
- 16 dicembre a Milano
- Il Natale 2023 di Pro RETT Ricerca
- Azzurra per il 5X1000 2023
- Pro RETT News #07 - marzo 2023
- Pasqua 2023 di Pro RETT Ricerca
- Diventa socio di Pro RETT Ricerca
- Il Natale 2022 di Pro RETT
- Due nuovi progetti nel 2022
- Riconfermato il Consiglio Direttivo
- Il 5x1000 a Pro RETT Ricerca
- Pasqua 2022
- L'Erbolario sostiene la ricerca scientifica
- Il mercatino di Natale 2021
- Dove c'è un bisogno, c'è un Lion
- Secondo incontro online...
- Incontro online...
- Uniti per accelerare...
- Il Natale 2020 di Pro RETT
- Campagna fotografica 2020
- Il Vostro 5x1000 a Pro RETT
- Pasqua 2020 con Pro RETT
- Le Voci di Rett
- 6th European Conference
Secondo incontro online di aggiornamento dai laboratori
1 Giugno 2021
Sabato 12 giugno, alle ore 15.00 su piattaforma Zoom, si terrà il secondo webinair di Pro RETT Ricerca con l'obiettivo di presentare i progetti e gli aggiornamenti sulle principali ricerche finanziate dall'associazione grazie al contributo di tutti i donatori.

Con la disponibilità del prof. Enrico Tongiorgi, Ordinario di Neurobiologia Cellulare e Neuroscienze Applicate e responsabile del Laboratorio di Neurobiologia Cellulare e dello Sviluppo al Dipartimento di Scienze della Vita presso l’Università degli Studi di Trieste, faremo il punto sulle ricerche in essere presso il suo laboratorio. Tra queste ci sarà anche lo studio sull'atrofia neuronale nelle pazienti Rett, presentato dalla dr.ssa Ottavia Roggero.
La partecipazione è gratuita ma l'iscrizione è obbligatoria. Se siete interessati a partecipare mandateci una mail all'indirizzo info@prorett.org specificando il vostro nominativo completo: verrete ricontattati per i dettagli.
Registratevi alla nostra newsletter per restare aggiornati e seguiteci sulla nostra pagina Facebook
Puoi effettuare la tua donazione online in totale sicurezza con la tua Carta di Credito o il tuo account PayPal.
Cliccando DONA ADESSO o il Logo PayPal qui sopra verrai reindirizzato al sito PayPal per concludere l'operazione.
- Margherita, la porta della speranza
- Pasqua 2024 di Pro RETT Ricerca
- Francesca Latella - 21 febbraio 2024
- 16 dicembre a Milano
- Il Natale 2023 di Pro RETT Ricerca
- Azzurra per il 5X1000 2023
- Pro RETT News #07 - marzo 2023
- Pasqua 2023 di Pro RETT Ricerca
- Diventa socio di Pro RETT Ricerca
- Il Natale 2022 di Pro RETT
- Due nuovi progetti nel 2022
- Riconfermato il Consiglio Direttivo
- Il 5x1000 a Pro RETT Ricerca
- Pasqua 2022
- L'Erbolario sostiene la ricerca scientifica
- Il mercatino di Natale 2021
- Dove c'è un bisogno, c'è un Lion
- Secondo incontro online...
- Incontro online...
- Uniti per accelerare...
- Il Natale 2020 di Pro RETT
- Campagna fotografica 2020
- Il Vostro 5x1000 a Pro RETT
- Pasqua 2020 con Pro RETT
- Le Voci di Rett
- 6th European Conference
Incontro online di aggiornamento dai laboratori
8 Maggio 2021
Sabato 15 maggio, alle ore 15.00, si terrà il primo evento online di Pro RETT Ricerca dedicato agli aggiornamenti dai laboratori impegnati nella ricerca di una cura per la sindrome di Rett.
Protagonista di questo incontro sarà la prof.ssa Nicoletta Landsberger, che ci offrirà una panoramica dei progetti di ricerca che sosteniamo presso i laboratori dell'Università degli Studi di Milano e in quelli del San Raffaele Rett Research Center.

La partecipazione è gratuita ma la registrazione è obbligatoria. Se siete interessati a partecipare mandateci una mail all'indirizzo info@prorett.org specificando il vostro nominativo completo: verrete ricontattati per i dettagli.
Abbiamo in programma altri due incontri: registratevi alla nostra newsletter per restare aggiornati.
Puoi effettuare la tua donazione online in totale sicurezza con la tua Carta di Credito o il tuo account PayPal.
Cliccando DONA ADESSO o il Logo PayPal qui sopra verrai reindirizzato al sito PayPal per concludere l'operazione.
- Margherita, la porta della speranza
- Pasqua 2024 di Pro RETT Ricerca
- Francesca Latella - 21 febbraio 2024
- 16 dicembre a Milano
- Il Natale 2023 di Pro RETT Ricerca
- Azzurra per il 5X1000 2023
- Pro RETT News #07 - marzo 2023
- Pasqua 2023 di Pro RETT Ricerca
- Diventa socio di Pro RETT Ricerca
- Il Natale 2022 di Pro RETT
- Due nuovi progetti nel 2022
- Riconfermato il Consiglio Direttivo
- Il 5x1000 a Pro RETT Ricerca
- Pasqua 2022
- L'Erbolario sostiene la ricerca scientifica
- Il mercatino di Natale 2021
- Dove c'è un bisogno, c'è un Lion
- Secondo incontro online...
- Incontro online...
- Uniti per accelerare...
- Il Natale 2020 di Pro RETT
- Campagna fotografica 2020
- Il Vostro 5x1000 a Pro RETT
- Pasqua 2020 con Pro RETT
- Le Voci di Rett
- 6th European Conference
Uniti per accelerare lo sviluppo di nuovi farmaci:
sinergia tra i laboratori di Milano e di Trieste grazie al supporto di Pro RETT Ricerca
16 Febbraio 2021
Negli ultimi mesi abbiamo lavorato molto sull'ottimizzazione dello stanziamento delle risorse economiche ai laboratori che sosteniamo grazie alle vostre donazioni e che coinvolgono un totale di 12 ricercatori, 1 tecnico di laboratorio, 2 studenti e 2 professori di facoltà. Il nostro obiettivo era, in primis, quello di fare tutto il possibile per accelerare i tempi della ricerca.
Se per alcuni progetti - come quello di terapia cellulare portato avanti dalla dr.ssa Angelisa Frasca presso l'Università degli Studi di Milano – il nostro margine di intervento è strettamente legato alle rigide tempistiche della sperimentazione, per i progetti di drug screening siamo riusciti a dare vita a una collaborazione tra il prof. Enrico Tongiorgi, del laboratorio Cellular and Developmental Neurobiology presso l’Università di Trieste, e la prof.ssa Nicoletta Landsberger, che coordina il lavoro nei due laboratori del San Raffaele Rett Research Center e del Dipartimento di Biotecnologie Mediche e Medicina Traslazionale presso l'Università degli Studi di Milano.
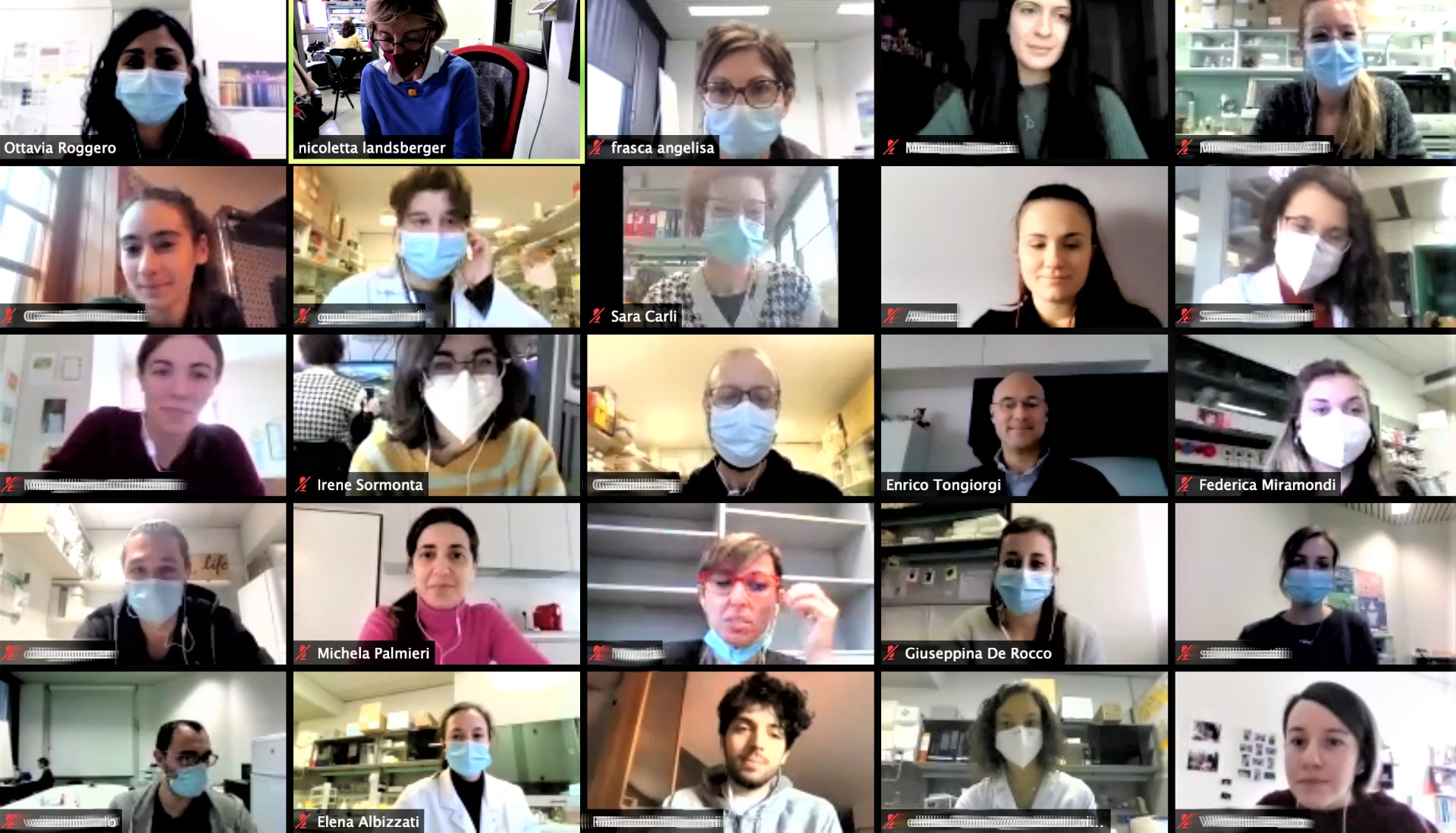
ll primo lab meeting tra l'Università degli Studi di Milano, il San Raffaele Rett Research Center e l'Università di Trieste.
Il 26 gennaio è stata quindi avviata una collaborazione tra i laboratori, già sostenuti da Pro RETT Ricerca, per sviluppare un progetto integrato finalizzato a far progredire più speditamente la sperimentazione di farmaci sui modelli pre-clinici (cellulari e animali) in preparazione di una possibile futura sperimentazione clinica. I due gruppi di ricerca fino a oggi hanno lavorato su binari paralleli, identificando varie decine di potenziali candidati farmaci nel laboratorio di Trieste e alcuni potenziali candidati nei laboratori di Milano.
Già dalle riunioni preliminari tenutesi a fine 2020, il Consiglio Direttivo di Pro RETT Ricerca aveva sentito l’urgenza di imprimere un’accelerazione alla sperimentazione di nuovi farmaci. La medesima necessità era stata avvertita anche dai due ricercatori che attualmente ricevono il nostro sostegno e che già dalla fine dell’anno precedente avevano iniziato a dialogare per trovare delle soluzioni efficaci ad abbreviare il tempo che intercorre tra i test di laboratorio e la sperimentazione clinica. Così, nella riunione telematica di gennaio 2021, al sollecito del Consiglio Direttivo in tal senso i due laboratori hanno prontamente risposto impegnandosi a sviluppare un progetto congiunto.
I laboratori di Trieste e di Milano stanno già lavorando al progetto mettendo a fattor comune le rispettive competenze. L’obiettivo comune è di effettuare nel più breve tempo possibile una serie di test in vitro selettivi e rigorosi per selezionare, come in una corsa a ostacoli, i farmaci più promettenti. Ogni laboratorio utilizzerà test ottimizzati nel proprio laboratorio utili a verificare le capacità di approcci farmacologici di migliorare o recuperare completamente i tipici difetti molecolari, morfologici o funzionali presentati dai neuroni Rett.
Il progetto prevede che i farmaci che riusciranno a superare con successo la maggioranza delle prove in vitro impostate dai ricercatori verranno poi testati in vivo per valutarne gli effetti su deficit del movimento, socializzazione, capacità cognitive, sensibilità sensoriale e sulla respirazione. Riguardo a quest’ultimo punto, Pro RETT Ricerca si è impegnata anche a garantire l'acquisto di un pletismografo; un apparecchio molto costoso (circa 30.000 euro) ma essenziale per misurare le apnee e che verrà dato in comodato d’uso gratuito al laboratorio della prof.ssa Landsberger presso UNIMI.
- Margherita, la porta della speranza
- Pasqua 2024 di Pro RETT Ricerca
- Francesca Latella - 21 febbraio 2024
- 16 dicembre a Milano
- Il Natale 2023 di Pro RETT Ricerca
- Azzurra per il 5X1000 2023
- Pro RETT News #07 - marzo 2023
- Pasqua 2023 di Pro RETT Ricerca
- Diventa socio di Pro RETT Ricerca
- Il Natale 2022 di Pro RETT
- Due nuovi progetti nel 2022
- Riconfermato il Consiglio Direttivo
- Il 5x1000 a Pro RETT Ricerca
- Pasqua 2022
- L'Erbolario sostiene la ricerca scientifica
- Il mercatino di Natale 2021
- Dove c'è un bisogno, c'è un Lion
- Secondo incontro online...
- Incontro online...
- Uniti per accelerare...
- Il Natale 2020 di Pro RETT
- Campagna fotografica 2020
- Il Vostro 5x1000 a Pro RETT
- Pasqua 2020 con Pro RETT
- Le Voci di Rett
- 6th European Conference
Il Natale 2020 di Pro RETT Ricerca
28 Ottobre 2020
Cari amici,
pur consapevoli delle difficoltà che stiamo vivendo a causa dell'emergenza sanitaria in corso, vogliamo rendervi partecipi della nostra proposta per arrivare a Natale sostenendo la ricerca di una cura per la sindrome di Rett.

Ad accompagnarci, quest'anno, ci sarà il volto della nostra Gemma, immortalata per noi dai fotografi di 6PMStudio di Roma con la collaborazione di mamma Elena da Siena.
Qui sotto troverete il nostro catalogo solidale (potrete scaricarlo anche qui), rivisto nella grafica e con una selezione di prodotti pensati per garantire a tutti i nostri sostenitori la qualità che contraddistingue le campagne di raccolta fondi di Pro RETT Ricerca.
Abbiamo attivato anche una sezione del sito chiamata "MERCATINO" (qui) dove potrete procedere in autonomia ed effettuare il pagamento tramite PayPal o bonifico bancario.
Per qualsiasi informazione scriveteci all'indirizzo segreteria@prorett.org: vi risponderemo (quasi) subito!

- Margherita, la porta della speranza
- Pasqua 2024 di Pro RETT Ricerca
- Francesca Latella - 21 febbraio 2024
- 16 dicembre a Milano
- Il Natale 2023 di Pro RETT Ricerca
- Azzurra per il 5X1000 2023
- Pro RETT News #07 - marzo 2023
- Pasqua 2023 di Pro RETT Ricerca
- Diventa socio di Pro RETT Ricerca
- Il Natale 2022 di Pro RETT
- Due nuovi progetti nel 2022
- Riconfermato il Consiglio Direttivo
- Il 5x1000 a Pro RETT Ricerca
- Pasqua 2022
- L'Erbolario sostiene la ricerca scientifica
- Il mercatino di Natale 2021
- Dove c'è un bisogno, c'è un Lion
- Secondo incontro online...
- Incontro online...
- Uniti per accelerare...
- Il Natale 2020 di Pro RETT
- Campagna fotografica 2020
- Il Vostro 5x1000 a Pro RETT
- Pasqua 2020 con Pro RETT
- Le Voci di Rett
- 6th European Conference
5X1000 2020: la nostra campagna fotografica
25 Luglio 2020
Si è conclusa la nostra campagna 2020 per promuovere il 5X1000 a Pro RETT Ricerca e vogliamo condividere alcune istantanee di quanto comunicato in queste settimane.
Grazie agli scatti dei fotografi Antonio Lorenzini e Cristina Corsi, sulle nostre pagine Facebook e Instagram abbiamo realizzato un racconto per immagini ma dobbiamo continuare a far sapere ad amici e parenti quanto sia importante destinare il 5X1000 a Pro RETT Ricerca se vogliamo accorciare i tempi per una cura.
La cura è possibile: fatelo sapere a tutti!

IO SONO MELLY
Melly ha 15 anni e abita a Salsomaggiore Terme, in Provincia di Parma. Nelle prossime settimane ci parlerà di sé, della sua famiglia e di alcuni momenti della sua giornata...
photo credit ©Antonio Lorenzini

IO SONO MELLY, io viaggio in bicicletta.
Questa è la bicicletta di Melly, lei è davanti sempre in prima linea; il nonno Valter la guida e pedala. Insieme possono percorrere le strade delle dolci colline di Salsomaggiore. Insieme possono volare...
photo credit ©Cristina Corsi

IO SONO MELLY, finalmente è l'ora di pranzo!
È un giorno di festa per Melly, oggi ci sono i nonni. Nonna Rita ha cucinato gli spaghetti con i pomodorini e il basilico. Mattia - suo fratello - è pronto a divorarli, mentre Melly rivolge alla nonna uno sguardo di gratitudine che va oltre ogni parola...
photo credit ©Antonio Lorenzini

IO SONO MELLY, ogni giorno un giretto nel pollaio!
È un momento importante per Melly entrare nel pollaio e distribuire il becchime alle galline che, impazienti, l’aspettano... Melly sa che in cambio troverà sempre un uovo, pronto per essere raccolto; un bianchissimo uovo solo per Melly...
photo credit ©Cristina Corsi

IO SONO MELLY, Mattia è mio fratello!
C'è un legame profondo tra Melly e Mattia. Non si può descrivere l'amore che provano l'uno per l'altra, dove la fragilità si unisce alla tenerezza e alla protezione. Un amore incondizionato, senza confini, che rassicura anche nei momenti più difficili della malattia...
photo credit ©Cristina Corsi

IO SONO MELLY... e anche io vado al bar!
Così, qualche volta Melly e la sua mamma vanno a prendere l'aperitivo al bar. Lo chiamano spritz, ma per Melly è Coca-Cola e patatine. Per Melly è una gioia andare al bar, incontrare tante persone che parlano con lei, perchè ormai la conoscono e forse anche l'aspettano...
photo credit ©Cristina Corsi

IO SONO MELLY... e vorrei prendere il treno.
La stazione di Salsomaggiore Terme è un luogo in cui Melly vuole spesso andare. Alcuni pomeriggi, quando l'ansia e la noia si fanno sentire, la mamma porta Melly a vedere i treni che arrivano, partono e sono pieni di persone che hanno tante cose da fare e luoghi da raggiungere...
photo credit ©Antonio Lorenzini

IO SONO MELLY e insieme camminiamo nel parco!
Melly percorre i sentieri del parco, mani sicure l'accompagnano. In questo parco siamo invitati tutti a entrare e a percorrere insieme a Melly le zone di ombra e di luce che gli alberi maestosi tracciano lungo i sentieri...
photo credit ©Antonio Lorenzini

IO SONO MELLY, il tuo 5x1000 è importante per me!
Oggi Melly finisce il suo racconto. Conclude con un sorriso e la richiesta di non mollare nel cercare una cura per la Rett. Per questo il tuo #5x1000 è importante per Melly, perché arriveremo a una cura solo se riusciremo ad avviare progetti di ricerca complessi che sappiano andare in profondità nella conoscenza della malattia... [continua a leggere].
photo credit ©Cristina Corsi
Il tuo passo verso di noi è importante
e solo con te faremo il salto che ci chiede Melly.
Firma la tua Dichiarazione dei Redditi nel riquadro a Sostegno delle
Organizzazioni Non Lucrative di Utilità Sociale e inserisci il Codice Fiscale
di Pro RETT Ricerca: 93043680201
SCOPRI DI PIU'
Puoi effettuare la tua donazione online in totale sicurezza con la tua Carta di Credito o il tuo account PayPal.
Cliccando DONA ADESSO o il Logo PayPal qui sopra verrai reindirizzato al sito PayPal per concludere l'operazione.
- Margherita, la porta della speranza
- Pasqua 2024 di Pro RETT Ricerca
- Francesca Latella - 21 febbraio 2024
- 16 dicembre a Milano
- Il Natale 2023 di Pro RETT Ricerca
- Azzurra per il 5X1000 2023
- Pro RETT News #07 - marzo 2023
- Pasqua 2023 di Pro RETT Ricerca
- Diventa socio di Pro RETT Ricerca
- Il Natale 2022 di Pro RETT
- Due nuovi progetti nel 2022
- Riconfermato il Consiglio Direttivo
- Il 5x1000 a Pro RETT Ricerca
- Pasqua 2022
- L'Erbolario sostiene la ricerca scientifica
- Il mercatino di Natale 2021
- Dove c'è un bisogno, c'è un Lion
- Secondo incontro online...
- Incontro online...
- Uniti per accelerare...
- Il Natale 2020 di Pro RETT
- Campagna fotografica 2020
- Il Vostro 5x1000 a Pro RETT
- Pasqua 2020 con Pro RETT
- Le Voci di Rett
- 6th European Conference
Il vostro 5x1000 può aiutarci a trovare una cura
31 Maggio 2021
In questi mesi, come milioni di italiani, sarete chiamati a depositare la dichiarazione dei redditi percepiti nel corso del 2021. In quell'occasione avrete la possibilità di scegliere a chi destinare il vostro 5x1000.
Il 5x1000 è una quota della vostra imposta IRPEF, quindi non vi verranno chiesti ulteriori oneri, che lo Stato ripartisce per dare sostegno a enti che svolgono attività socialmente rilevanti, ossia che lavorano per l'interesse proprio della pluralità e della collettività. Il cittadino contribuente può effettuare una scelta precisa, come per l’8 e il 2x1000, mentre la quota di coloro che non indicano un codice fiscale specifico viene ripartita proporzionalmente a seconda delle scelte sottoscritte dagli altri.
Noi di Pro RETT Ricerca siamo genitori volontari, non percepiamo stipendio ma lavoriamo, tutti i giorni, per trovare i fondi necessari a finanziare la ricerca scientifica sulla sindrome di Rett nella speranza di arrivare presto a una cura per le nostre figlie.
Se state leggendo queste pagine significa che non abbiamo bisogno di spiegarvi cosa comporti essere ammalati della sindrome di Rett. Siamo una piccola onlus, mandiamo poche comunicazioni dirette e solo a contatti selezionati, quindi sappiamo per certo che sapete già tutto ciò di cui avete bisogno per capire quanto sia importante, per noi e le nostre figlie, che scegliate Pro RETT Ricerca come destinataria di quelle tasse che avete già pagato.
Come fare?
Cercate nel CUD rilasciato dal datore di lavoro o dall’INPS la pagina dedicata alla scelte, con un campo per la firma e uno per il Codice Fiscale della realtà che volete sostenere. Nella sezione 5x1000, in alto a sinistra, mettete la vostra firma nella casella "Sostegno degli Enti del Terzo settore [...] e delle Onlus iscritte all'anagrafe" e inserite il nostro Codice Fiscale:
93043680201
Nel caso non foste chiamati a compilare la dichiarazione dei redditi avrete comunque la possibilità di destinare il vostro 5x1000 a Pro RETT Ricerca consegnando quella pagina, debitamente compilata e in busta chiusa con l’indicazione del vostro codice fiscale, presso un Ufficio Postale o un CAF abilitato alla trasmissione telematica (ma ormai lo sono quasi tutti).
Oggi, però, vi chiediamo uno sforzo maggiore. Molte persone scelgono a chi destinare il 5x1000 solo per poter chiudere la compilazione della dichiarazione dei redditi alla svelta e non pensarci più. Ecco: portate la nostra voce tra i vostri colleghi, fate giungere la nostra storia ai vostri familiari e agli amici che incontrate dopo il lavoro. Raccontate loro di quello che facciamo; spiegategli che quel 5x1000 “donato” a Pro RETT Ricerca viene utilizzato per pagare lo stipendio dei ricercatori che studiano questa malattia rara e che altrimenti non potrebbero farlo in Italia, per rinnovare la strumentazione e acquistare materiali di consumo nei laboratori dell'Università degli Studi di Milano e presso il San Raffaele Rett Research Center.
Aiutateci a spargere la voce; aiutateci a farlo sapere a tutti.
---
Ringraziamo la fotografa Cristina Corsi per lo scatto alla nostra Beatrice. Cristina lavora insieme al fotografo Antonio Lorenzini per un progetto sulla sindrome di Rett di cui vi parleremo presto.
Puoi effettuare la tua donazione online in totale sicurezza con la tua Carta di Credito o il tuo account PayPal.
Cliccando DONA ADESSO o il Logo PayPal qui sopra verrai reindirizzato al sito PayPal per concludere l'operazione.
- Margherita, la porta della speranza
- Pasqua 2024 di Pro RETT Ricerca
- Francesca Latella - 21 febbraio 2024
- 16 dicembre a Milano
- Il Natale 2023 di Pro RETT Ricerca
- Azzurra per il 5X1000 2023
- Pro RETT News #07 - marzo 2023
- Pasqua 2023 di Pro RETT Ricerca
- Diventa socio di Pro RETT Ricerca
- Il Natale 2022 di Pro RETT
- Due nuovi progetti nel 2022
- Riconfermato il Consiglio Direttivo
- Il 5x1000 a Pro RETT Ricerca
- Pasqua 2022
- L'Erbolario sostiene la ricerca scientifica
- Il mercatino di Natale 2021
- Dove c'è un bisogno, c'è un Lion
- Secondo incontro online...
- Incontro online...
- Uniti per accelerare...
- Il Natale 2020 di Pro RETT
- Campagna fotografica 2020
- Il Vostro 5x1000 a Pro RETT
- Pasqua 2020 con Pro RETT
- Le Voci di Rett
- 6th European Conference
Pasqua 2020 con Pro RETT Ricerca
Acquista le uova di Pasqua di Pro RETT Ricerca!
7 Marzo 2020
È ai nastri di partenza la nostra campagna di raccolta fondi per il periodo pasquale, con la distribuzione di migliaia di uova al cioccolato (fondente o al latte da 350 grammi) a tutte le nostre famiglie di riferimento sul territorio.
Ciascun uovo solidale è caratterizzato da un collarino realizzato per esplicitare il valore sociale di questo prodotto e spiegare la destinazione dei fondi raccolti. Se avete delle famiglie di riferimento a cui rivolgervi per l'acquisto di piccoli quantitativi potete farlo già da oggi. Per ordini importanti potete invece mandarci una mail all'indirizzo segreteria@prorett.org chiedendo tutte le informazioni che riterrete opportune oppure cliccando sul link 'Contattaci' in questa pagina.

La Sindrome di Rett è una malattia genetica che colpisce prevalentemente le bambine, che crescono apparentemente sane per i primi mesi di vita (6-24) poi subentra una rapida regressione e in breve tempo perdono tutte le abilità acquisite: la capacità di camminare, di parlare, di usare le mani e si arresta lo sviluppo psicomotorio. La causa della malattia è la mutazione del gene MECP2 che è coinvolto in altre patologie neuropsichiatriche come l'autismo, la disapìbilità intellettuale e la depressione. Nel 2007 è stato dimostrato che la sindrome di Rett è REVERSIBILE su modelli animali e può diventare una malattia CURABILE. Ecco perchè continuare a SOSTENERE la ricerca significa dare una SPERANZA di vita migliore alle bambine colpite dalla sindrome di Rett in tutto il mondo.
LE UOVA DI PRORETT RICERCA SONO ARTIGIANALI, DISPONIBILI AL CIOCCOLATO AL LATTE O FONDENTE DA 350 gr/cad. CON UN CONTRIBUTO DI 10 EURO.
Per tutti gli aggiornamenti seguiteci sulla nostra pagina Facebook!
- Margherita, la porta della speranza
- Pasqua 2024 di Pro RETT Ricerca
- Francesca Latella - 21 febbraio 2024
- 16 dicembre a Milano
- Il Natale 2023 di Pro RETT Ricerca
- Azzurra per il 5X1000 2023
- Pro RETT News #07 - marzo 2023
- Pasqua 2023 di Pro RETT Ricerca
- Diventa socio di Pro RETT Ricerca
- Il Natale 2022 di Pro RETT
- Due nuovi progetti nel 2022
- Riconfermato il Consiglio Direttivo
- Il 5x1000 a Pro RETT Ricerca
- Pasqua 2022
- L'Erbolario sostiene la ricerca scientifica
- Il mercatino di Natale 2021
- Dove c'è un bisogno, c'è un Lion
- Secondo incontro online...
- Incontro online...
- Uniti per accelerare...
- Il Natale 2020 di Pro RETT
- Campagna fotografica 2020
- Il Vostro 5x1000 a Pro RETT
- Pasqua 2020 con Pro RETT
- Le Voci di Rett
- 6th European Conference
Fate sentire la vostra voce!
18 Febbraio 2020
Care famiglie,
dopo tanto lavoro, con grande impegno di Orietta Mariotti - membro del Consiglio Direttivo di Pro RETT Ricerca - siamo felici di annunciare ufficialmente la nostra collaborazione con le principali associazioni che si occupano della sindrome di Rett nel mondo per la realizzazione di un questionario unico e, finora, mai tentato.

Abbiamo lavorato (e lavoreremo) al fianco di Rettsyndrome.org, Rett UK, Reverse Rett, Rett Syndrome Association of Australia e Rett Deutschland e.V.. Queste associazioni, come la stessa Pro RETT Ricerca, sono state selezionate tra le più importanti parental advocacy association del panorama internazionale per essere i vettori di una mappatura globale di tutte le famiglie Rett: delle loro difficoltà, dei loro bisogni (anche economici).

Il questionario fornirà informazioni e dati importantissimi per misurare l’impatto fisico, emotivo e finanziario della sindrome di Rett sulle bambine, sulle famiglie, sugli operatori sanitari e la comunità Rett nel suo complesso.
Le informazioni raccolte grazie a questo questionario saranno utilizzate per aiutare a trovare farmaci, programmi e servizi destinati a coloro che sono affetti da questa malattia.
Per accedere alle domande e completare il questionario (Clicca qui), che potrà essere salvato in ogni momento e che, quindi, potrà essere portato a termine in più sessioni, avrete bisogno di un codice di accesso. Questo codice ve lo fornirà la nostra segreteria, quindi vi chiediamo di scriverci una mail (segreteria@prorett.org) facendone specifica richiesta.

Non è ancora finita: è stato predisposto un questionario anche per il personale medico e sanitario, con domande dediate e un rimborso spese per il tempo impiegato nel rispondere ai quesiti. Con questa premessa, vi chiediamo di segnalarci (esclusivamente via mail) eventuali medici o professionisti con cui collaborate costantemente, indicandoci il loro indirizzo di posta elettronica, nome e cognome, così da poterli raggiungere mettendovi in copia.
Qualora possibile, vi chiediamo di condividere sui vostri canali di comunicazione questa opportunità, siano essi profili Facebook o contatti su Whatsapp.
Nei prossimi giorni torneremo da voi con alcuni aggiornamenti.
Un cordiale saluto,
la segreteria.
Puoi effettuare la tua donazione online in totale sicurezza con la tua Carta di Credito o il tuo account PayPal.
Cliccando DONA ADESSO o il Logo PayPal qui sopra verrai reindirizzato al sito PayPal per concludere l'operazione.
- Margherita, la porta della speranza
- Pasqua 2024 di Pro RETT Ricerca
- Francesca Latella - 21 febbraio 2024
- 16 dicembre a Milano
- Il Natale 2023 di Pro RETT Ricerca
- Azzurra per il 5X1000 2023
- Pro RETT News #07 - marzo 2023
- Pasqua 2023 di Pro RETT Ricerca
- Diventa socio di Pro RETT Ricerca
- Il Natale 2022 di Pro RETT
- Due nuovi progetti nel 2022
- Riconfermato il Consiglio Direttivo
- Il 5x1000 a Pro RETT Ricerca
- Pasqua 2022
- L'Erbolario sostiene la ricerca scientifica
- Il mercatino di Natale 2021
- Dove c'è un bisogno, c'è un Lion
- Secondo incontro online...
- Incontro online...
- Uniti per accelerare...
- Il Natale 2020 di Pro RETT
- Campagna fotografica 2020
- Il Vostro 5x1000 a Pro RETT
- Pasqua 2020 con Pro RETT
- Le Voci di Rett
- 6th European Conference
TERAPIE TRATTAMENTI E CURE
al 6th European Rett Syndrome Conference Finlandia Settembre 2019
a cura di Enrico Tongiorgi,
Professore associato presso l'Università di Trieste
2 Dicembre 2019
A settembre si è svolta in Finlandia la 6th European Rett Syndrome Conference. Al convegno hanno partecipato le associazioni dei parenti di ventisette paesi che rappresentano nel complesso oltre seimila pazienti affette dalla sindrome di Rett. Era presente anche il Vicepresidente di Pro RETT Ricerca Michele Baruffaldi e lo vedete in una delle foto accanto al nostro poster.
L’associazione Rett Finland, che ha organizzato il convegno assieme alla Rett Syndrome Europe (RSE), ha selezionato un numero ristretto di ricercatori e medici con l’intento di avere relazioni focalizzate principalmente sullo stato dell’arte degli interventi clinici attualmente in sviluppo. A questo proposito, James Eubanks dell’Università di Toronto (Canada) ha precisato che gli interventi clinici si distinguono in terapie, trattamenti e cure. Le “Terapie” mirano a ottenere dei miglioramenti nella qualità di vita delle pazienti e delle loro famiglie attraverso per esempio fisioterapia, ippoterapia, nuoto, musico-terapia e apprendimento tramite computer. La differenza tra “Trattamenti” e “Cure”, nel caso della sindrome di Rett è piuttosto chiara: mentre i trattamenti utilizzano farmaci e integratori alimentari per ridurre alcuni sintomi (per esempio farmaci antiepilettici o farmaci per la respirazione o integratori antiossidanti), le cure mirano invece a ripristinare in parte o del tutto le funzioni del gene MeCP2 tramite terapia genica con vari approcci, oppure mediante somministrazione del MeCP2 sotto forma di proteina sintetica.
Terapie
Iniziamo con le terapie per le bambine, ragazze e adulte Rett. Nel corso del convegno sono state presentate esperienze di terapia fisica volte a rinforzare la muscolatura necessaria per sostenere la postura (in piedi e seduta), l’abilità di camminare e l’uso delle mani. In particolare, Lotan Meier della Ariel University (Israele) ha evidenziato come il terapista e le famiglie debbano partire dal presupposto che le ragazze sono in grado di percepire molto di più di quanto possono rispondere. Il primo passo della terapia fisica deve essere quindi quello di stabilire un forte rapporto emotivo con la paziente Rett prima dell’inizio dell’intervento e di mantenerlo costante durante tutta la terapia scegliendo modalità di comunicazione e oggetti graditi alla paziente, creando un ambiente che infonda un senso di serenità e sicurezza. Per raggiungere questo obiettivo familiari ed educatori possono e devono essere coinvolti, quantomeno fornendo informazioni utili sulle preferenze della paziente ed essendo a conoscenza del percorso terapeutico. Il passo successivo per il terapista è di individuare dei fattori motivanti quali musica, animali, gioco che aumentino il grado di cooperazione della paziente nell’intraprendere la terapia. Lotan Meier ha raccomandato che gli operatori, con l’aiuto dei familiari, prestino grande attenzione alle espressioni facciali delle ragazze in trattamento per stabilire se l’intervento è più o meno gradito, incoraggiando ed utilizzando nella terapia le iniziative individuali delle ragazze. Sempre tenendo presente che le reazioni emotive e fisiche possono a volta arrivare anche minuti dopo lo stimolo, in quanto le ragazze Rett hanno bisogno dei propri tempi e del rispetto dei loro momenti di assenza ma anche di ansia, paura e rifiuto o desiderio di divertimento. Infine, il terzo passo è quello di stabilire un programma di terapia fisica costante nel tempo con ripetizioni continue di esercizi semplici per minimizzare la necessità di un apprendimento fino ad arrivare ad una esecuzione spontanea degli esercizi, possibilmente con un incremento nell’intensità.
Con Pro RETT Ricerca all'Advanced Therapies Science Meeting di Berlino
Ed è proprio questo che Angelisa e io abbiamo deciso di partecipare al primo Advanced Therapies Science Meeting (www.restore-horizon.eu) che si terrà a Berlino il 25 e 26 novembre 2019. Si tratta di un meeting voluto e organizzato da eminenti esperti di ricerca di base, clinica e biotecnologica sostenuti da più di trecento rappresentanti del mondo dell’industria, delle organizzazioni non-profit e dei sostenitori dei pazienti derivanti da oltre ventisei paesi nel mondo, con lo scopo di fare il punto e scoprire gli ultimi aggiornamenti sui progressi nel campo delle Terapie Avanzate. Ci sarà data ampia possibilità anche di discutere, insieme agli altri ricercatori, di quali siano le principali sfide da affrontare e come accelerare l’inserimento nella clinica di queste terapie. L’importanza dell’evento è testimoniata dal discorso di apertura del congresso che verrà tenuto dal premio Nobel per la Chimica Ada Yonath, attualmente direttore dell’Istituto Scientifico Weizmann di Israele. Il programma sarà fitto e siamo felici di informarvi che anche noi parteciperemo direttamente, come protagoniste con uno spazio dedicato al nostro lavoro, perché il nostro gruppo di ricerca è stato selezionato per presentare i risultati ottenuti sino a ora nella sessione dedicata ai “nuovi target e nuove indicazioni”.
Di qualità di vita delle pazienti e delle famiglie Rett ha parlato in modo più approfondito Aglaia Vignoli dell’Università di Milano, dirigente medico presso la neuropsicologia infantile dell’Ospedale Santi Carlo e Paolo di Milano. In particolare, ha evidenziato la necessità di utilizzare dei questionari specifici per valutare quali aspetti della malattia delle figlie Rett influenzano la qualità di vita e la salute dei genitori. Dalle indagini condotte negli ultimi anni in vari paesi, risulta che le difficoltà di alimentazione, il numero ridotto delle ore di sonno, i problemi comportamentali e le crisi epilettiche delle ragazze Rett sono percepite dai genitori come gli aspetti che hanno maggiore impatto sulla qualità di vita dell’intera famiglia. Per entrambi i genitori sono invece risultati come attenuatori dello stress la possibilità di avere un lavoro fuori casa almeno part-time, la stabilità economica, il supporto sociale, i servizi scolastici e medico-assistenziali. Gli aspetti della qualità di vita che risultano colpire di più i genitori delle ragazze Rett riguardano la salute mentale (per es. fenomeni di ansia e depressione), le relazioni sociali (per es. riduzione delle relazioni, difficoltà di spiegare e far accettare socialmente la malattia della figlia), il livello di vitalità in generale e di salute complessiva (per es. frequenza malattie infettive, dolori fisici).

Infine, un aspetto particolarmente rilevante per medici e famiglie riguarda la difficoltà di comunicare con le ragazze Rett. A questo proposito, le nuove tecnologie di rilevazione dei movimenti oculari associate a vari metodi di comunicazione mediante computer offrono oggi delle straordinarie opportunità. Su questo tema vi sono state varie presentazioni. Anna Amato e Anne Berger del Dipartimento di logopedia di Berlino, hanno parlato di Augmentative and Alternative Communication. Questo sistema si basa su immagini proposte su tessere di cartone o su computer (eventualmente guidato dai movimenti oculari) che riportano dei simboli molto semplici che consentono di facilitare la comprensione di un rapporto di causa-effetto tra due eventi e di far comunicare da parte delle ragazze Rett una scelta SI-NO. Questo sistema consente di annunciare l’inizio di una attività (mangiare, dormire, lavarsi, etc..) e di far scegliere alle pazienti Rett un’attività o la modalità per svolgere una attività.
Trattamenti
Per quanto riguarda i trattamenti, i vari interventi al convegno hanno sottolineato come siano in corso numerosi studi clinici per la sperimentazione di farmaci volti alla risoluzione o quanto meno all’attenuazione di sintomi specifici. Jeffrey Neuls della Vanderbilt University (USA) ha parlato dei risultati della sperimentazione clinica con Mecasermin e con Trofinetide che mostrano effetti modesti e solo ad alto dosaggio. Un nuovo studio di fase 2 con il farmaco anestetico Ketamina, condotto in 6 siti negli USA grazie al supporto dalla fondazione Rett Syndrome Research Trust, sta valutando la sicurezza e tollerabilità di singole dosi. In questo studio, tuttora in corso, si valuteranno anche gli effetti sulla respirazione e su scale di valutazione globale da parte dei clinici e delle famiglie. L’azienda ARCH sta reclutando pazienti negli USA, UK, Spagna e Italia per una sperimentazione clinica del Cannabidiolo per 24 settimane. Come risultati ci si attende un miglioramento nelle scale di valutazione Rett Syndrome Behavior Questionnaire e Clinical Global Impression. L’azienda ANAVEX ha riportato i primi risultati ottenuti dallo studio clinico con la somministrazione per 7 settimane di 5 mg/giorno di Blarcamesine, un farmaco che attiva il recettore Sigma-1 e il recettore muscarinico dell’acetilcolina. I dati sui primi 6 pazienti sono risultati incoraggianti secondo Walter Kaufmann, coinvolto nello studio, anche considerando che ha riguardato solo le pazienti più grandi (18-36 anni di età). Lo studio clinico STAR promosso in USA, UK e Italia dalla NEWRON con Sarizotan ha come target primario il miglioramento delle crisi di apnea. I risultati dello studio sono attesi entro la fine del 2019.
Enrico Tongiorgi dell’Università di Trieste, ha riportato i risultati di uno studio retrospettivo sulla somministrazione di Mirtazapina in pazienti adulte in cura da Joussef Hayek e Claudio De Felice presso l’Ospedale Le Scotte di Siena. Mirtazapina, un antidepressivo e ansiolitico, era stato somministrato ad 11 pazienti di età tra 18 e 40 anni per la sua nota capacità di migliorare i disturbi dell’umore, l’ansia e il sonno. Grazie ad uno studio comparato con gli effetti sui topi Tongiorgi, Hayek e De Felice hanno identificato nelle pazienti un effetto positivo proprio nei 6 comportamenti che dal recente studio di Walter Kaufmann e collaboratori risultano compromettere maggiormente la qualità della vita delle pazienti Rett e delle loro famiglie.
Nel suo intervento, Enrico Tongiorgi ha anche descritto la strategia del progetto di drug screening sostenuto da Pro RETT Ricerca per l’identificazione di nuovi farmaci capaci di contrastare l’atrofia dei neuroni Rett. Nella prima fase di screening alcune decine di sostanze sono risultate efficaci in un modello cellulare della sindrome di Rett e al momento sono in corso gli esperimenti di verifica di questi primi incoraggianti risultati.

Cure
Infine, come già sottolineato, le cure con farmaci che permettono di riattivare il gene MeCP2, o di “leggere” il gene superando le mutazioni di stop, sono ancora nella fase di sperimentazione su cellule e modelli animali nei quali si sta soprattutto valutando la potenziale grande tossicità di queste sostanze. Per la terapia genica, sono stati messi a punto dei virus modificati per esprimere il gene MeCP2 nelle cellule delle pazienti, rimpiazzando l’allele mutato. Tuttavia esistono delle serie preoccupazioni che questi virus possano causare la sindrome di duplicazione genica quando espressi nelle cellule che portano l’allele normale del MecP2 dove il gene virale si aggiunge al gene normalmente funzionante, raddoppiando la dose di proteina espressa. Christophe Roux dell’Università di Marsiglia (Francia) ha condotto esperimenti di inoculazione di un virus che esprime il fattore neurotrofico BDNF (Brain-derived neurotrophic factor) nei topi con delezione del gene MeCP2. I risultati preliminari riportano un miglioramento della sopravvivenza e una riduzione delle apnee, tuttavia la strada per arrivare ad una terapia genica appare ancora molto lunga e non priva di potenziali gravi rischi.
- Margherita, la porta della speranza
- Pasqua 2024 di Pro RETT Ricerca
- Francesca Latella - 21 febbraio 2024
- 16 dicembre a Milano
- Il Natale 2023 di Pro RETT Ricerca
- Azzurra per il 5X1000 2023
- Pro RETT News #07 - marzo 2023
- Pasqua 2023 di Pro RETT Ricerca
- Diventa socio di Pro RETT Ricerca
- Il Natale 2022 di Pro RETT
- Due nuovi progetti nel 2022
- Riconfermato il Consiglio Direttivo
- Il 5x1000 a Pro RETT Ricerca
- Pasqua 2022
- L'Erbolario sostiene la ricerca scientifica
- Il mercatino di Natale 2021
- Dove c'è un bisogno, c'è un Lion
- Secondo incontro online...
- Incontro online...
- Uniti per accelerare...
- Il Natale 2020 di Pro RETT
- Campagna fotografica 2020
- Il Vostro 5x1000 a Pro RETT
- Pasqua 2020 con Pro RETT
- Le Voci di Rett
- 6th European Conference



- © 2024 Pro RETT Ricerca ONLUS |
- Web Design: web-communication.eu